

Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences
- PMID: 35148677
- PMCID: PMC8832810
- DOI: 10.1186/s12864-022-08358-2
Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences
Abstract
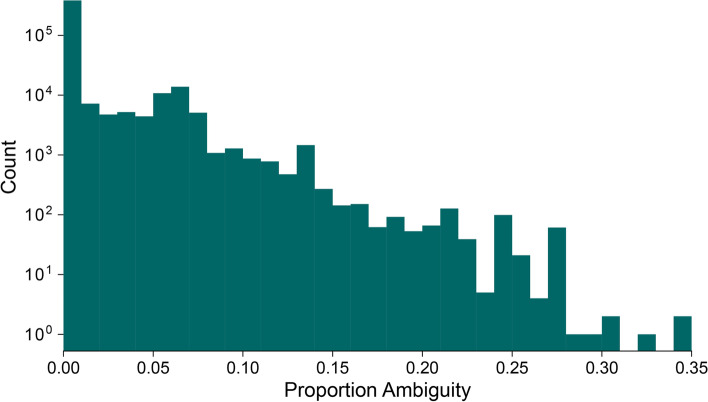
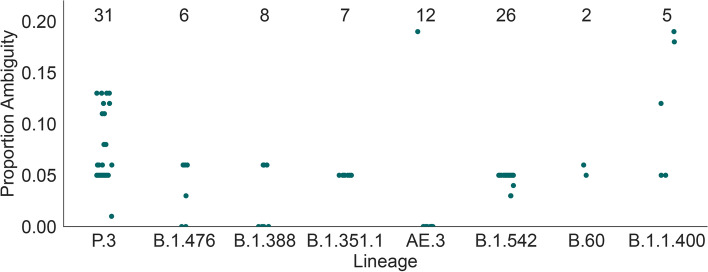
Background: More than 2 million SARS-CoV-2 genome sequences have been generated and shared since the start of the COVID-19 pandemic and constitute a vital information source that informs outbreak control, disease surveillance, and public health policy. The Pango dynamic nomenclature is a popular system for classifying and naming genetically-distinct lineages of SARS-CoV-2, including variants of concern, and is based on the analysis of complete or near-complete virus genomes. However, for several reasons, nucleotide sequences may be generated that cover only the spike gene of SARS-CoV-2. It is therefore important to understand how much information about Pango lineage status is contained in spike-only nucleotide sequences. Here we explore how Pango lineages might be reliably designated and assigned to spike-only nucleotide sequences. We survey the genetic diversity of such sequences, and investigate the information they contain about Pango lineage status.
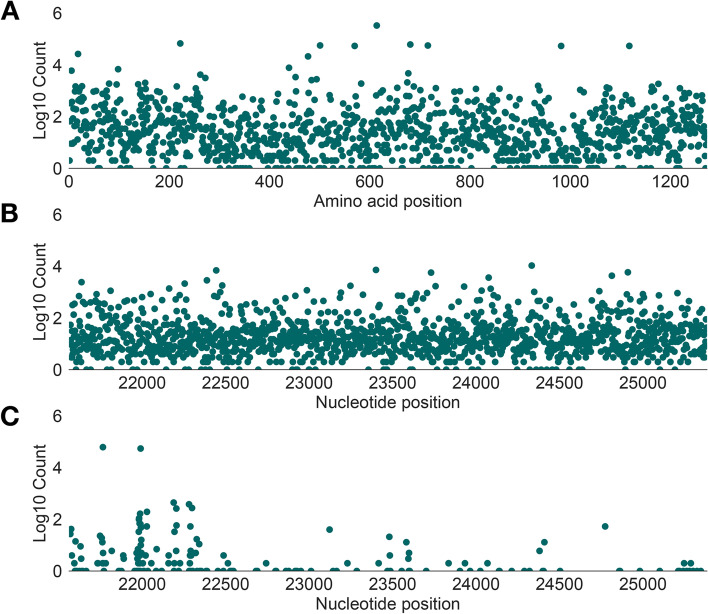
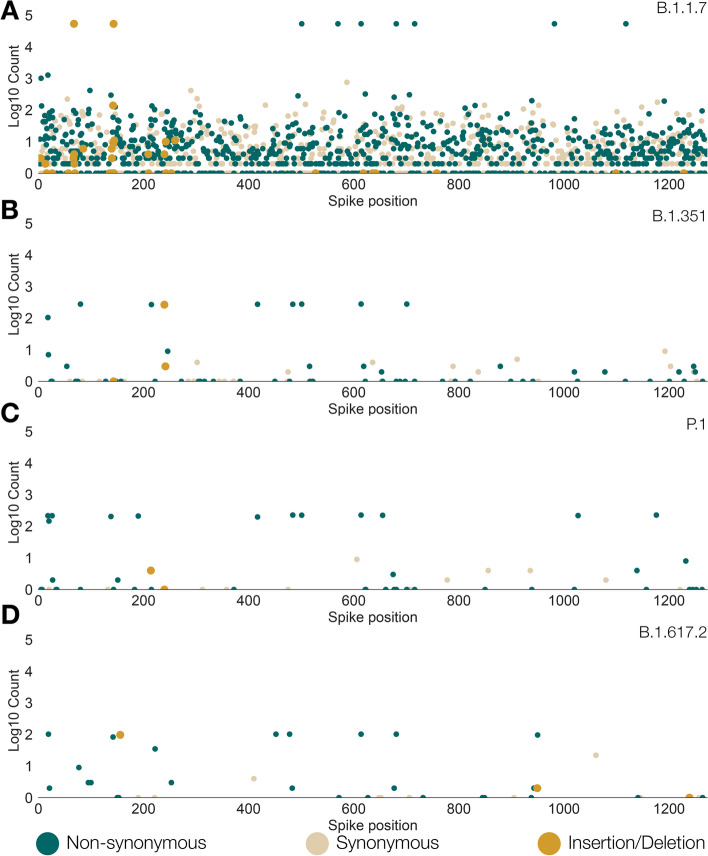
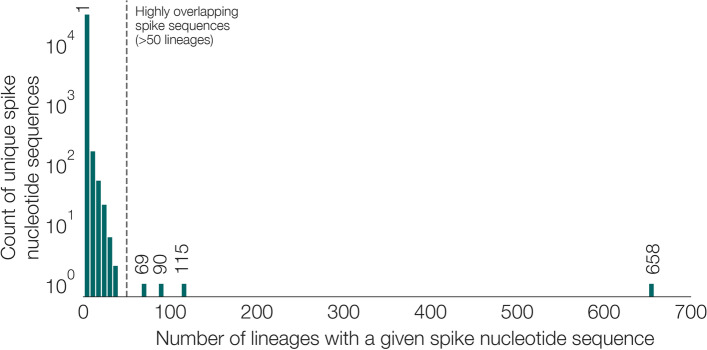
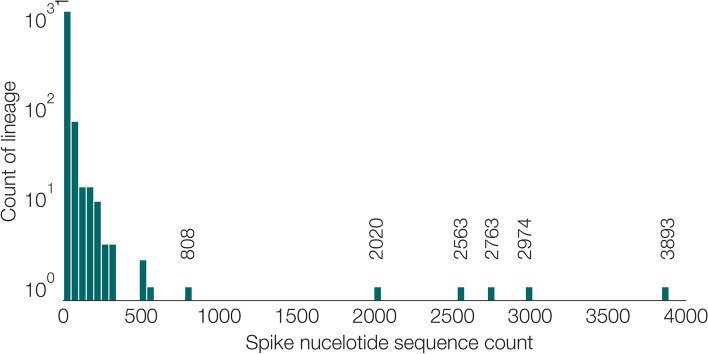
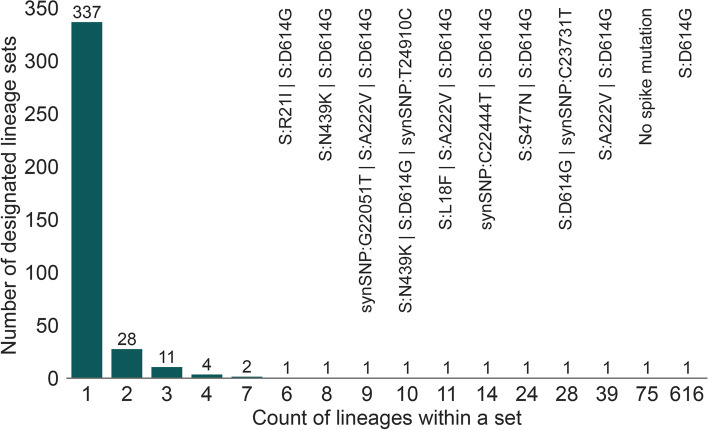
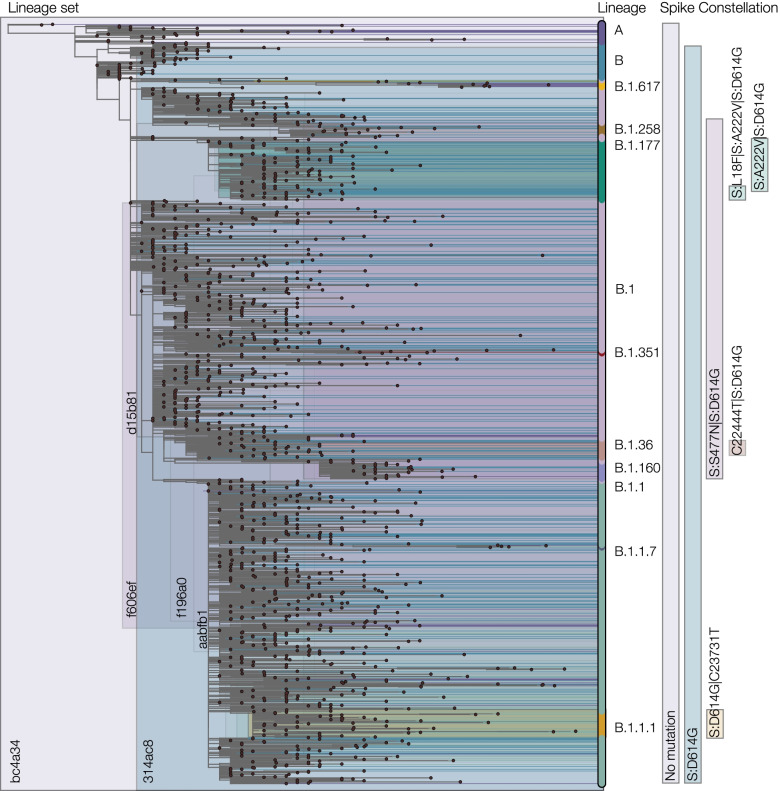
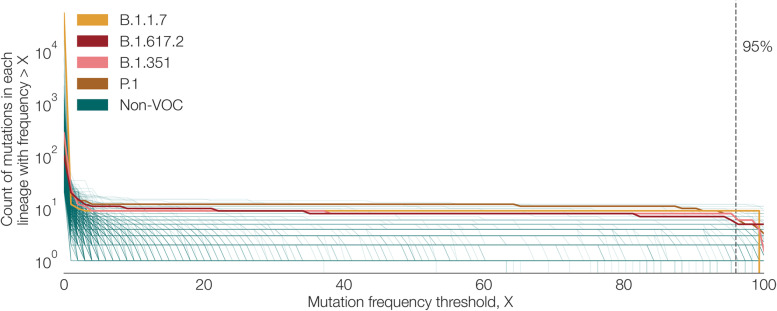
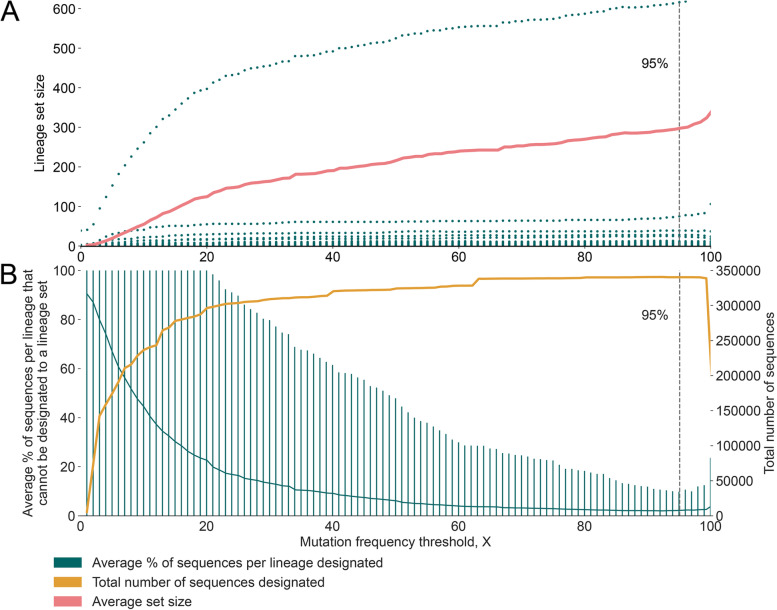
Results: Although many lineages, including the main variants of concern, can be identified clearly using spike-only sequences, some spike-only sequences are shared among tens or hundreds of Pango lineages. To facilitate the classification of SARS-CoV-2 lineages using subgenomic sequences we introduce the notion of designating such sequences to a "lineage set", which represents the range of Pango lineages that are consistent with the observed mutations in a given spike sequence.
Conclusions: We find that many lineages, including the main variants-of-concern, can be reliably identified by spike alone and we define lineage-sets to represent the lineage precision that can be achieved using spike-only nucleotide sequences. These data provide a foundation for the development of software tools that can assign newly-generated spike nucleotide sequences to Pango lineage sets.
Keywords: Genomic surveillance; Lineage; Pango; SARS-CoV-2; Spike.
© 2022. The Author(s).
Conflict of interest statement
MEA and EJK are employees of AstraZeneca and own stock.
Figures
References
-
- Jackson B, et al. Generation and transmission of inter-lineage recombinants in the SARS-CoV-2 pandemic. medRxiv. 2021. 10.1101/2021.06.18.21258689.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
