

cfTrack: A Method of Exome-Wide Mutation Analysis of Cell-free DNA to Simultaneously Monitor the Full Spectrum of Cancer Treatment Outcomes Including MRD, Recurrence, and Evolution
- PMID: 35149536
- PMCID: PMC9126584
- DOI: 10.1158/1078-0432.CCR-21-1242
cfTrack: A Method of Exome-Wide Mutation Analysis of Cell-free DNA to Simultaneously Monitor the Full Spectrum of Cancer Treatment Outcomes Including MRD, Recurrence, and Evolution
Erratum in
-
Correction: cfTrack: A Method of Exome-Wide Mutation Analysis of Cell-free DNA to Simultaneously Monitor the Full Spectrum of Cancer Treatment Outcomes Including MRD, Recurrence, and Evolution.Clin Cancer Res. 2026 Mar 16;32(6):1181. doi: 10.1158/1078-0432.CCR-26-0515. Clin Cancer Res. 2026. PMID: 41834509 Free PMC article. No abstract available.
Abstract
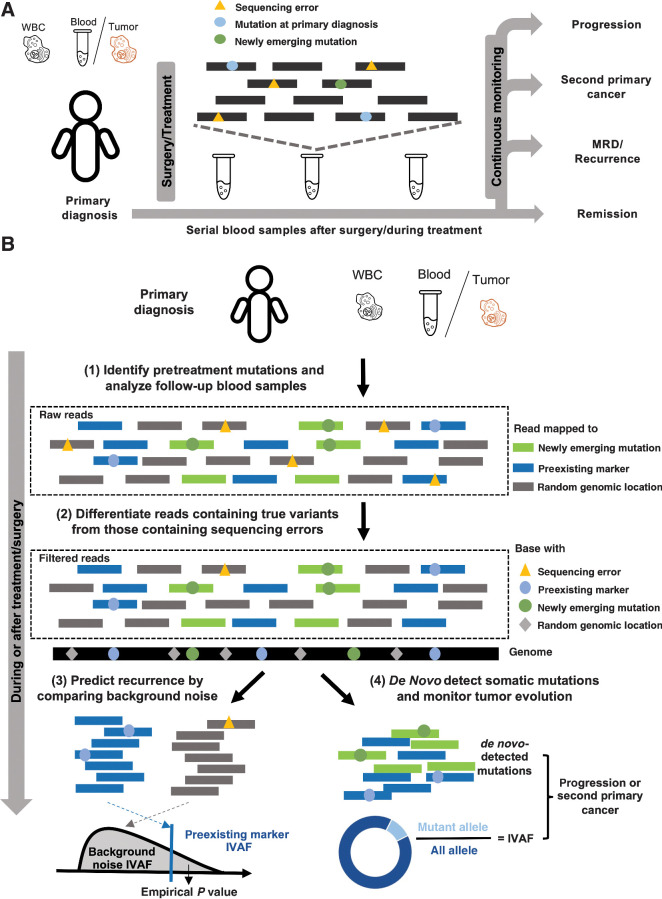
Purpose: Cell-free DNA (cfDNA) offers a noninvasive approach to monitor cancer. Here we develop a method using whole-exome sequencing (WES) of cfDNA for simultaneously monitoring the full spectrum of cancer treatment outcomes, including minimal residual disease (MRD), recurrence, evolution, and second primary cancers.
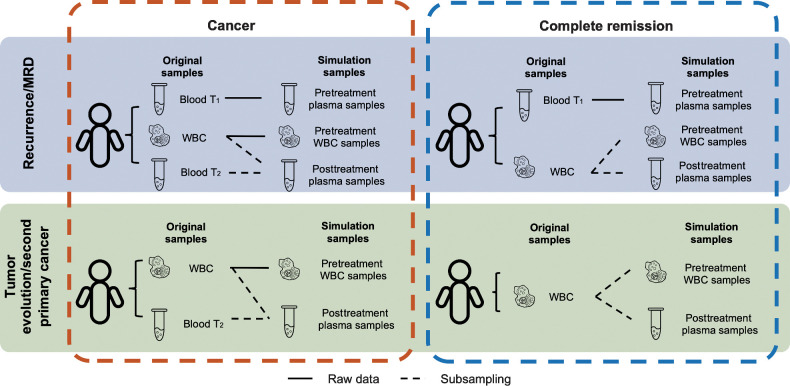
Experimental design: Three simulation datasets were generated from 26 patients with cancer to benchmark the detection performance of MRD/recurrence and second primary cancers. For further validation, cfDNA samples (n = 76) from patients with cancer (n = 35) with six different cancer types were used for performance validation during various treatments.
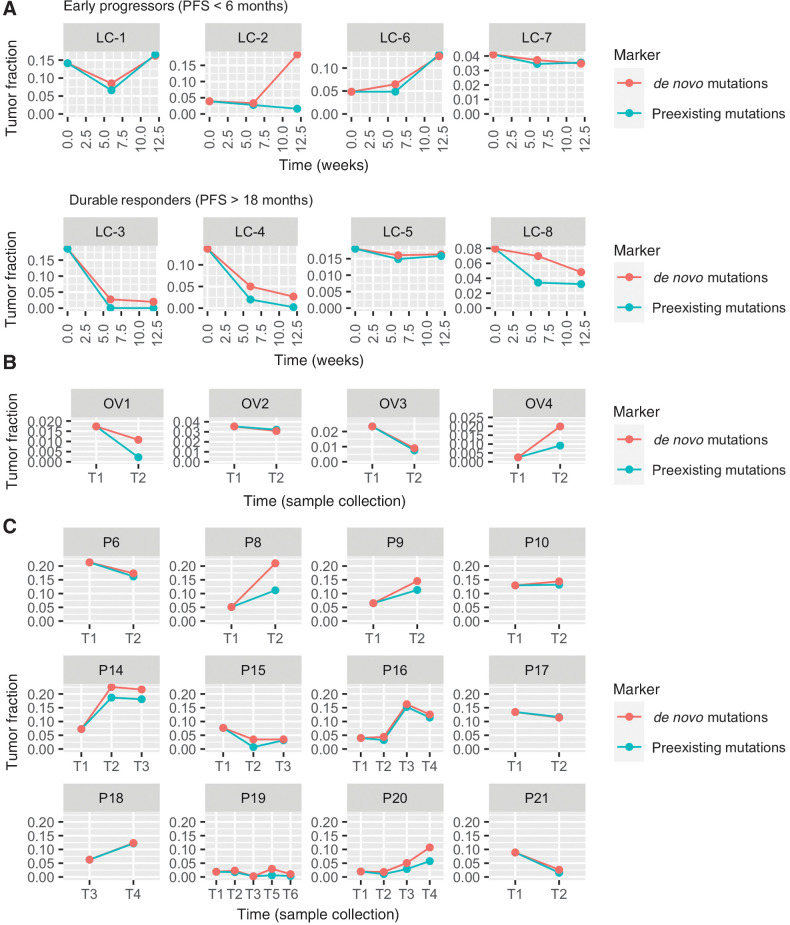
Results: We present a cfDNA-based cancer monitoring method, named cfTrack. Taking advantage of the broad genome coverage of WES data, cfTrack can sensitively detect MRD and cancer recurrence by integrating signals across known clonal tumor mutations of a patient. In addition, cfTrack detects tumor evolution and second primary cancers by de novo identifying emerging tumor mutations. A series of machine learning and statistical denoising techniques are applied to enhance the detection power. On the simulation data, cfTrack achieved an average AUC of 99% on the validation dataset and 100% on the independent dataset in detecting recurrence in samples with tumor fractions ≥0.05%. In addition, cfTrack yielded an average AUC of 88% in detecting second primary cancers in samples with tumor fractions ≥0.2%. On real data, cfTrack accurately monitors tumor evolution during treatment, which cannot be accomplished by previous methods.
Conclusions: Our results demonstrated that cfTrack can sensitively and specifically monitor the full spectrum of cancer treatment outcomes using exome-wide mutation analysis of cfDNA.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
![Figure 3. Performance of cancer recurrence and MRD detection using the simulation data. The area under the ROC curve (AUC) of the MRD/recurrence detection on the validation dataset (A) and the independent dataset (C) with different tumor fractions and sequencing depths. The sensitivity and specificity with different tumor fractions and sequencing depth on the validation dataset (B) and the independent dataset (D). Supplementary Figure S6A–S6D is the zoom-in of A–D at low tumor fraction ranging from 0% to 0.2%. E, AUCs of MRD/recurrence detection with and without error suppression (ES) on the validation dataset at 200× depth with different tumor fractions. F, The sensitivity and specificity of MRD/recurrence detection with and without error suppression on the validation dataset at 200× depth with different tumor fractions. In A, C, and E, the dots indicate the average AUC, and the vertical bars indicate average ± SD of the AUC (see Material and Methods). In B, D, and F, the dots show the average sensitivity using a cut-off P value = 0.05 for the background noise distribution; the vertical bars indicate average ± SD of the sensitivity; the specificity is shown in the legend in the format of [average specificity, (average − SD, average + SD)]. The solid lines show the smoothed performance fitted with logit functions.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/033d/9365349/c24101e9435c/1841fig3.jpg)
![Figure 4. Performance of second primary cancer detection with the simulation data. A, AUC of the in silico spike-in samples with different tumor fractions at 200× sequencing depth. The dots indicate the average AUC, and the vertical bars indicate average ± SD of the AUC (see Material and Methods). B, The sensitivity and specificity in the in silico spike-in samples with different tumor fractions at 200× sequencing depth. The dots show the average sensitivity using a cutoff of the 95th percentile of prediction scores from the remission samples in the training data; the vertical bars indicate average ± SD of the sensitivity; the specificity is shown in the text in the format of [average specificity, (average − SD, average + SD)]. The solid lines show the smoothed performance fitted with a logit function.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/033d/9365349/083722ffb67c/1841fig4.jpg)
References
-
- Kumar SK, Rajkumar SV. The current status of minimal residual disease assessment in myeloma. Leukemia 2014;28:239–40. - PubMed
