

De Novo and Dominantly Inherited SPTAN1 Mutations Cause Spastic Paraplegia and Cerebellar Ataxia
- PMID: 35150594
- PMCID: PMC9232883
- DOI: 10.1002/mds.28959
De Novo and Dominantly Inherited SPTAN1 Mutations Cause Spastic Paraplegia and Cerebellar Ataxia
Abstract
Background: Pathogenic variants in SPTAN1 have been linked to a remarkably broad phenotypical spectrum. Clinical presentations include epileptic syndromes, intellectual disability, and hereditary motor neuropathy.
Objectives: We investigated the role of SPTAN1 variants in rare neurological disorders such as ataxia and spastic paraplegia.
Methods: We screened 10,000 NGS datasets across two international consortia and one local database, indicative of the level of international collaboration currently required to identify genes causative for rare disease. We performed in silico modeling of the identified SPTAN1 variants.
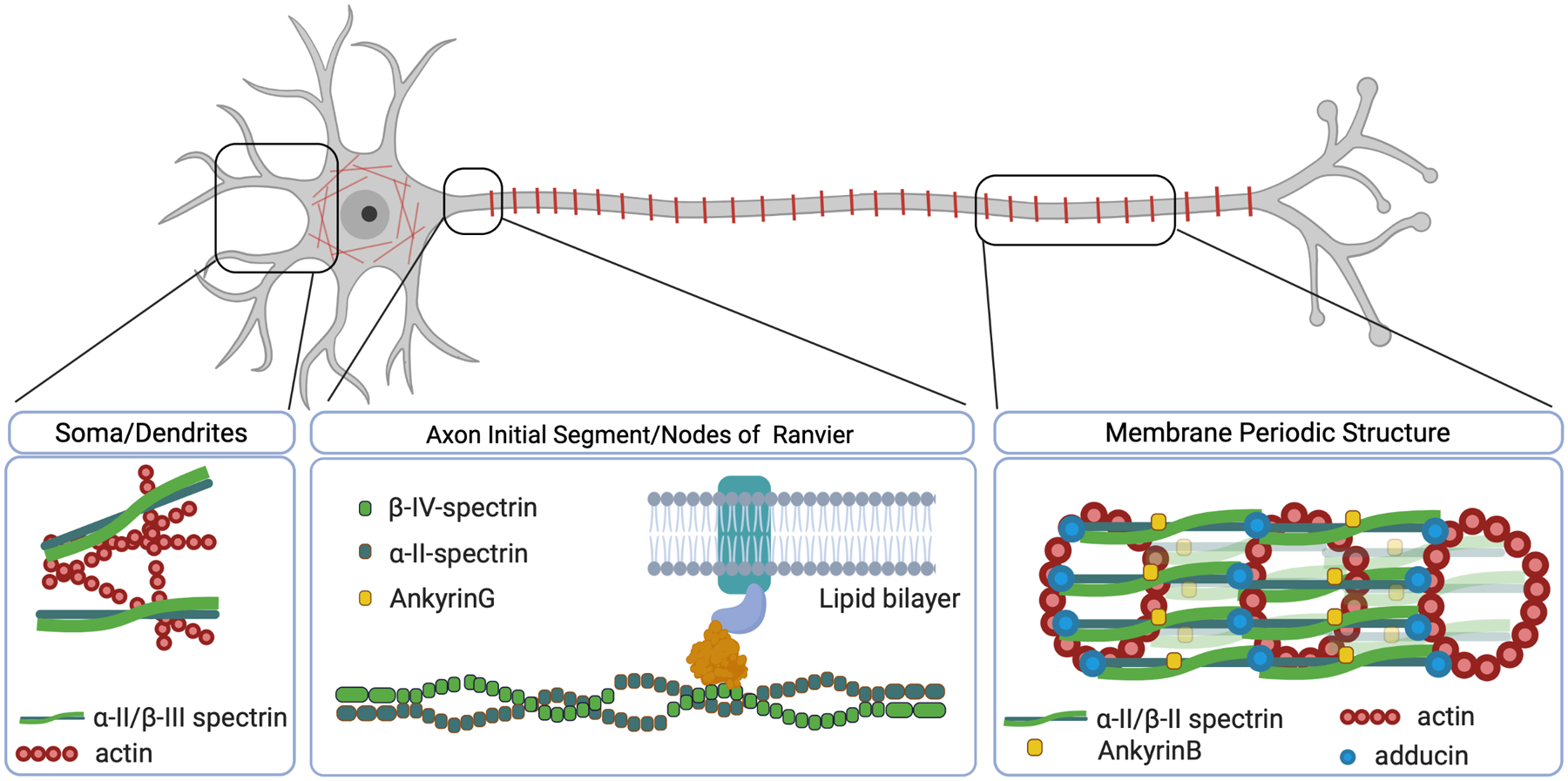
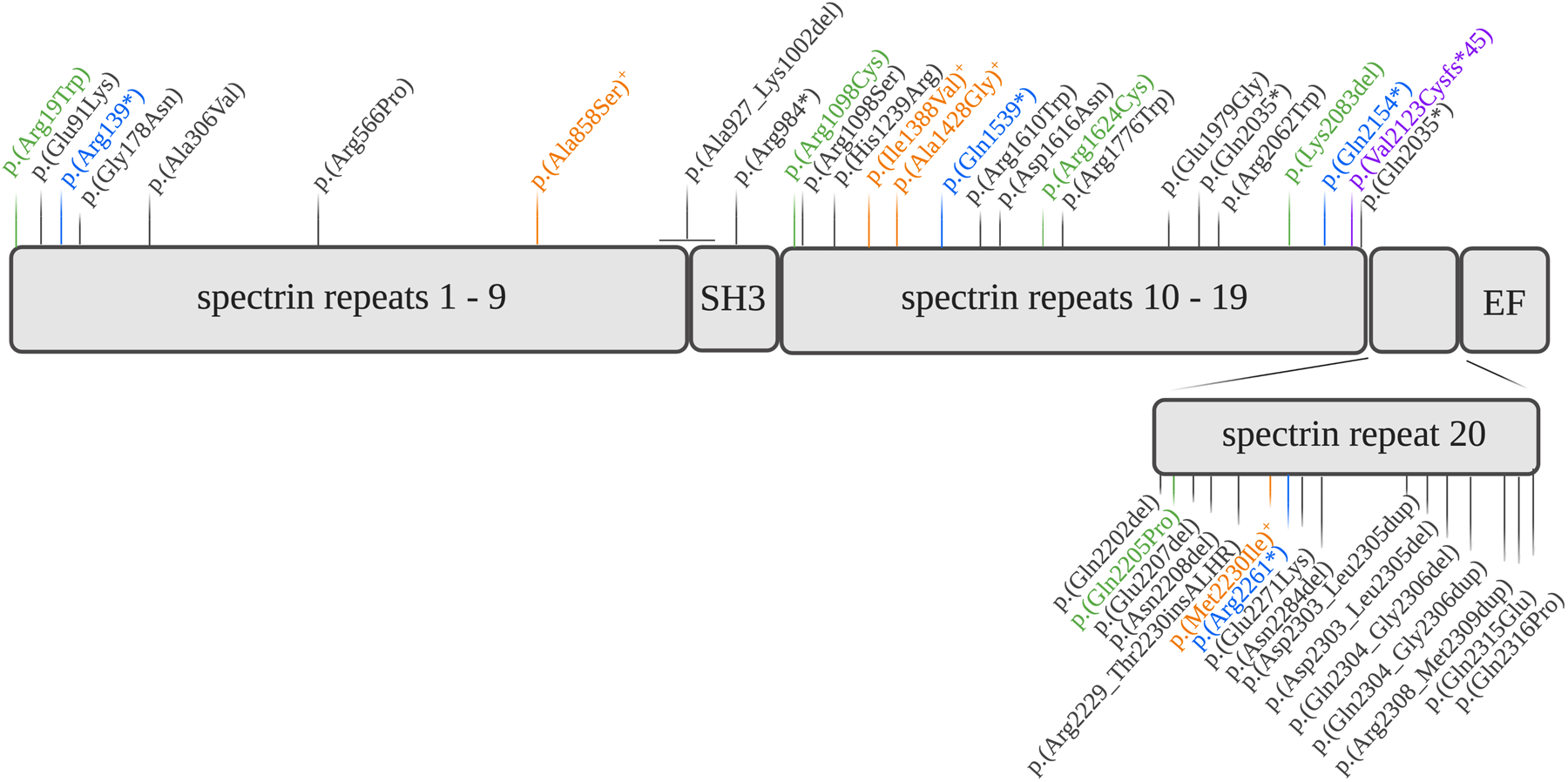
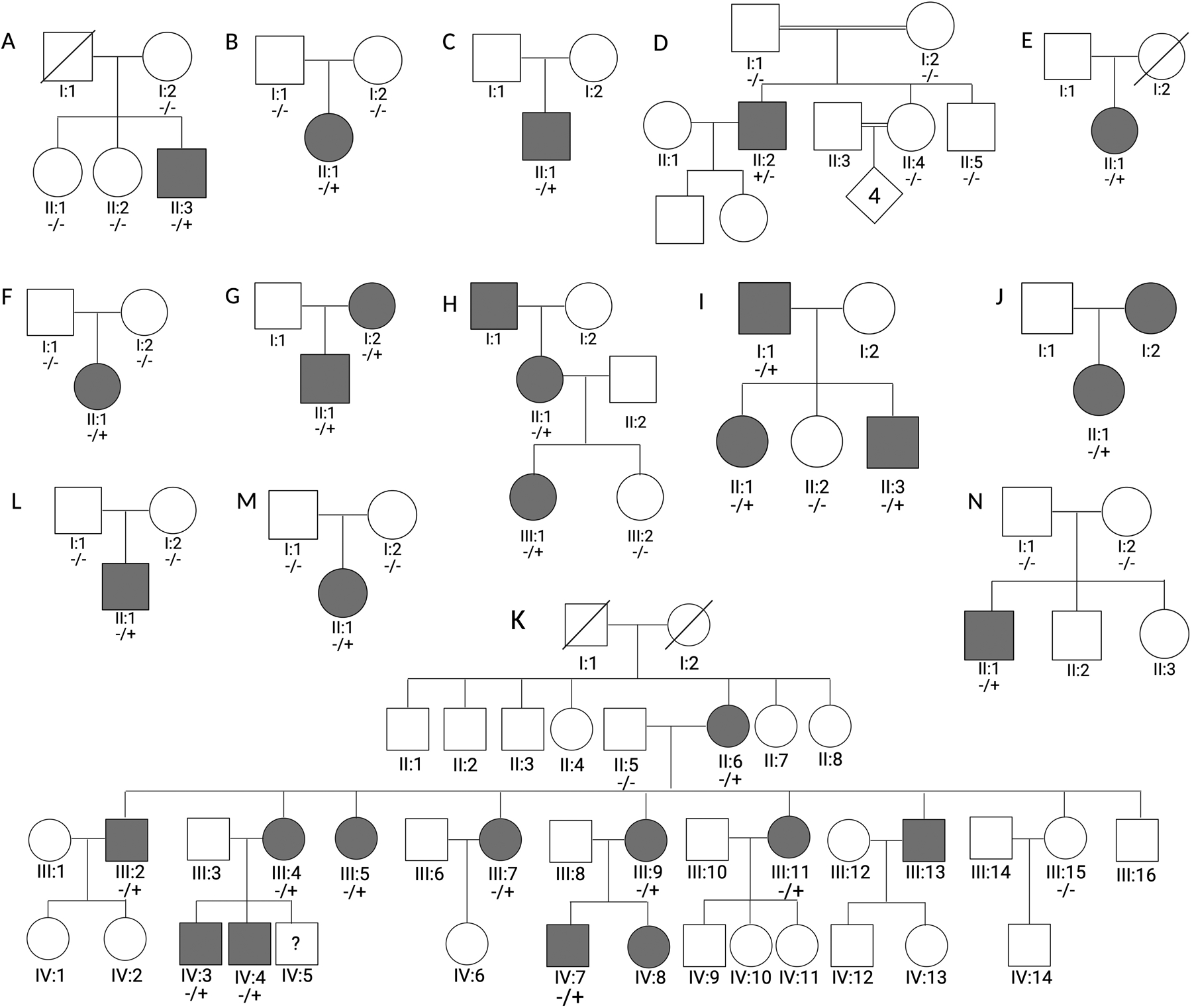
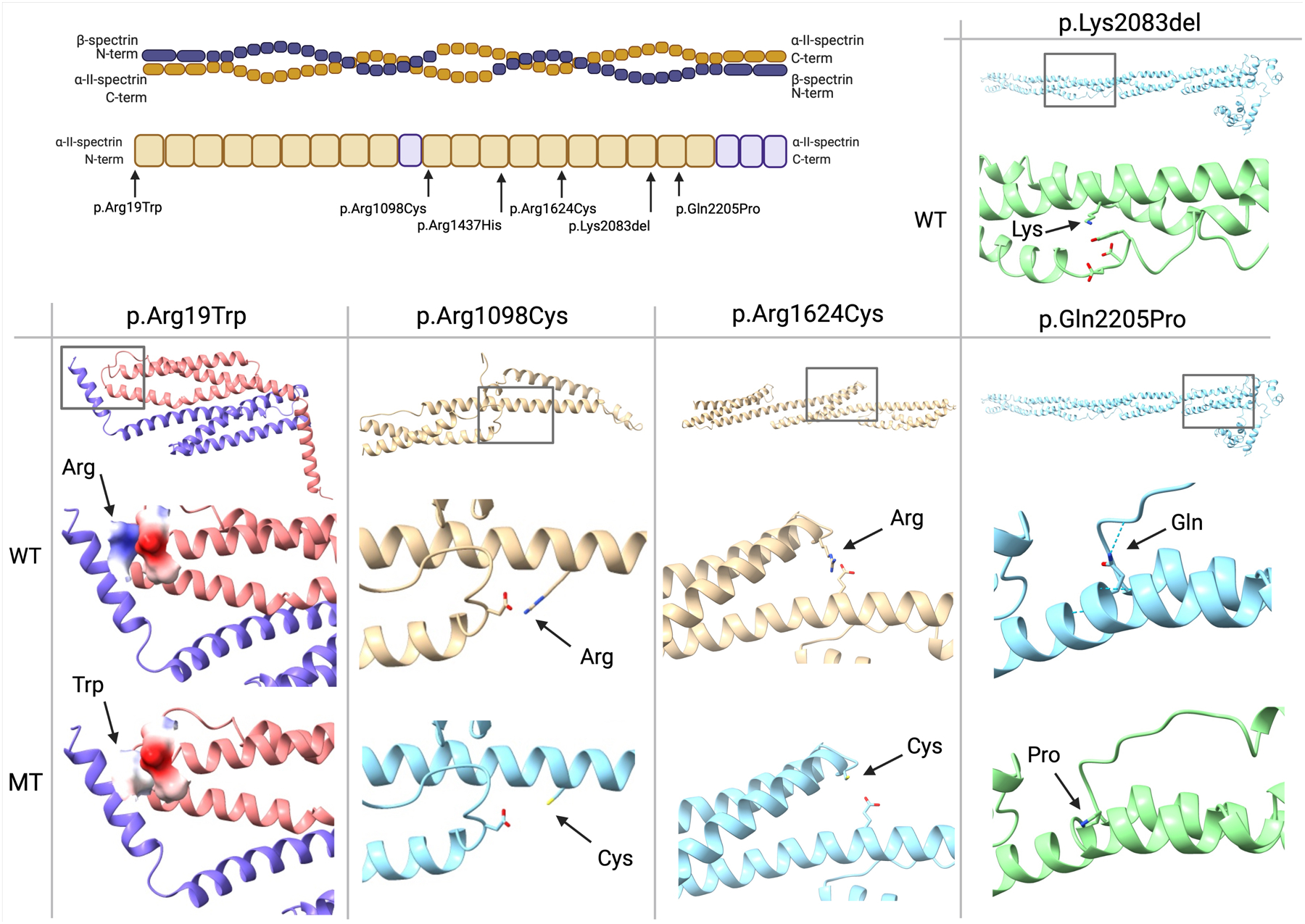
Results: We describe 22 patients from 14 families with five novel SPTAN1 variants. Of six patients with cerebellar ataxia, four carry a de novo SPTAN1 variant and two show a sporadic inheritance. In this group, one variant (p.Lys2083del) is recurrent in four patients. Two patients have novel de novo missense mutations (p.Arg1098Cys, p.Arg1624Cys) associated with cerebellar ataxia, in one patient accompanied by intellectual disability and epilepsy. We furthermore report a recurrent missense mutation (p.Arg19Trp) in 15 patients with spastic paraplegia from seven families with a dominant inheritance pattern in four and a de novo origin in one case. One further patient carrying a de novo missense mutation (p.Gln2205Pro) has a complex spastic ataxic phenotype. Through protein modeling we show that mutated amino acids are located at crucial interlinking positions, interconnecting the three-helix bundle of a spectrin repeat.
Conclusions: We show that SPTAN1 is a relevant candidate gene for ataxia and spastic paraplegia. We suggest that for the mutations identified in this study, disruption of the interlinking of spectrin helices could be a key feature of the pathomechanism. © 2022 International Parkinson and Movement Disorder Society.
Keywords: ataxia; next-generation sequencing; rare diseases; spastic paraplegia; spectrin.
© 2022 International Parkinson and Movement Disorder Society.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
