

A mouse model of inherited choline kinase β-deficiency presents with specific cardiac abnormalities and a predisposition to arrhythmia
- PMID: 35151687
- PMCID: PMC8913350
- DOI: 10.1016/j.jbc.2022.101716
A mouse model of inherited choline kinase β-deficiency presents with specific cardiac abnormalities and a predisposition to arrhythmia
Abstract
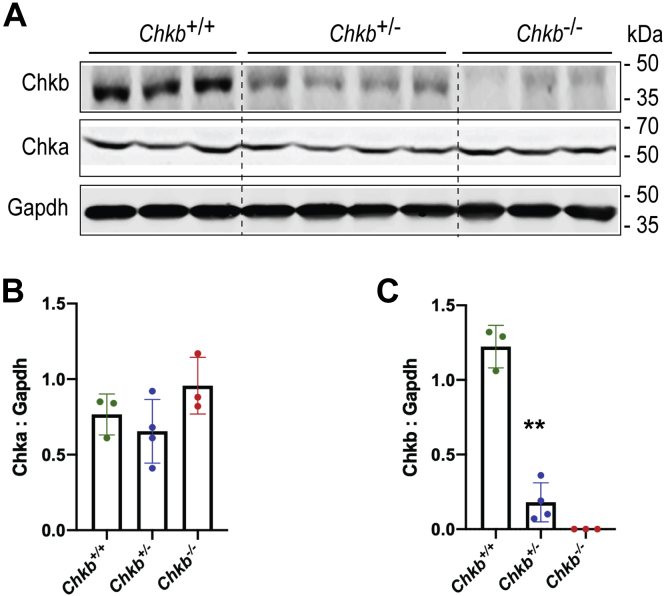
The CHKB gene encodes choline kinase β, which catalyzes the first step in the biosynthetic pathway for the major phospholipid phosphatidylcholine. Homozygous loss-of-function variants in human CHKB are associated with a congenital muscular dystrophy. Dilated cardiomyopathy is present in some CHKB patients and can cause heart failure and death. Mechanisms underlying a cardiac phenotype due to decreased CHKB levels are not well characterized. We determined that there is cardiac hypertrophy in Chkb-/- mice along with a decrease in left ventricle size, internal diameter, and stroke volume compared with wildtype and Chkb+/- mice. Unlike wildtype mice, 60% of the Chkb+/- and all Chkb-/- mice tested displayed arrhythmic events when challenged with isoproterenol. Lipidomic analysis revealed that the major change in lipid level in Chkb+/- and Chkb-/- hearts was an increase in the arrhythmogenic lipid acylcarnitine. An increase in acylcarnitine level is also associated with a defect in the ability of mitochondria to use fatty acids for energy and we observed that mitochondria from Chkb-/- hearts had abnormal cristae and inefficient electron transport chain activity. Atrial natriuretic peptide (ANP) is a hormone produced by the heart that protects against the development of heart failure including ventricular conduction defects. We determined that there was a decrease in expression of ANP, its receptor NPRA, as well as ventricular conduction system markers in Chkb+/- and Chkb-/- mice.
Keywords: acylcarnitine; cardiac muscle; heart disease; lipid; metabolism; muscular dystrophy; phosphatidylcholine.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures







References
-
- Molnar P., Hickman J.J. Modeling of action potential generation in NG108-15 cells. Methods Mol. Biol. 2014;1183:253–261. - PubMed
-
- Whited A.M., Johs A. The interactions of peripheral membrane proteins with biological membranes. Chem. Phys. Lipids. 2015;192:51–59. - PubMed
-
- Vance J.E. Phospholipid synthesis and transport in mammalian cells. Traffic. 2015;16:1–18. - PubMed
-
- Sher R.B., Aoyama C., Huebsch K.A., Ji S., Kerner J., Yang Y., Frankel W.N., Hoppel C.L., Wood P.A., Vance D.E., Cox G.A. A rostrocaudal muscular dystrophy caused by a defect in choline kinase beta, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 2006;281:4938–4948. - PubMed
-
- Mitsuhashi S., Ohkuma A., Talim B., Karahashi M., Koumura T., Aoyama C., Kurihara M., Quinlivan R., Sewry C., Mitsuhashi H., Goto K., Koksal B., Kale G., Ikeda K., Taguchi R., et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am. J. Hum. Genet. 2011;88:845–851. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
