

Accurate MHC Motif Deconvolution of Immunopeptidomics Data Reveals a Significant Contribution of DRB3, 4 and 5 to the Total DR Immunopeptidome
- PMID: 35154160
- PMCID: PMC8826445
- DOI: 10.3389/fimmu.2022.835454
Accurate MHC Motif Deconvolution of Immunopeptidomics Data Reveals a Significant Contribution of DRB3, 4 and 5 to the Total DR Immunopeptidome
Abstract
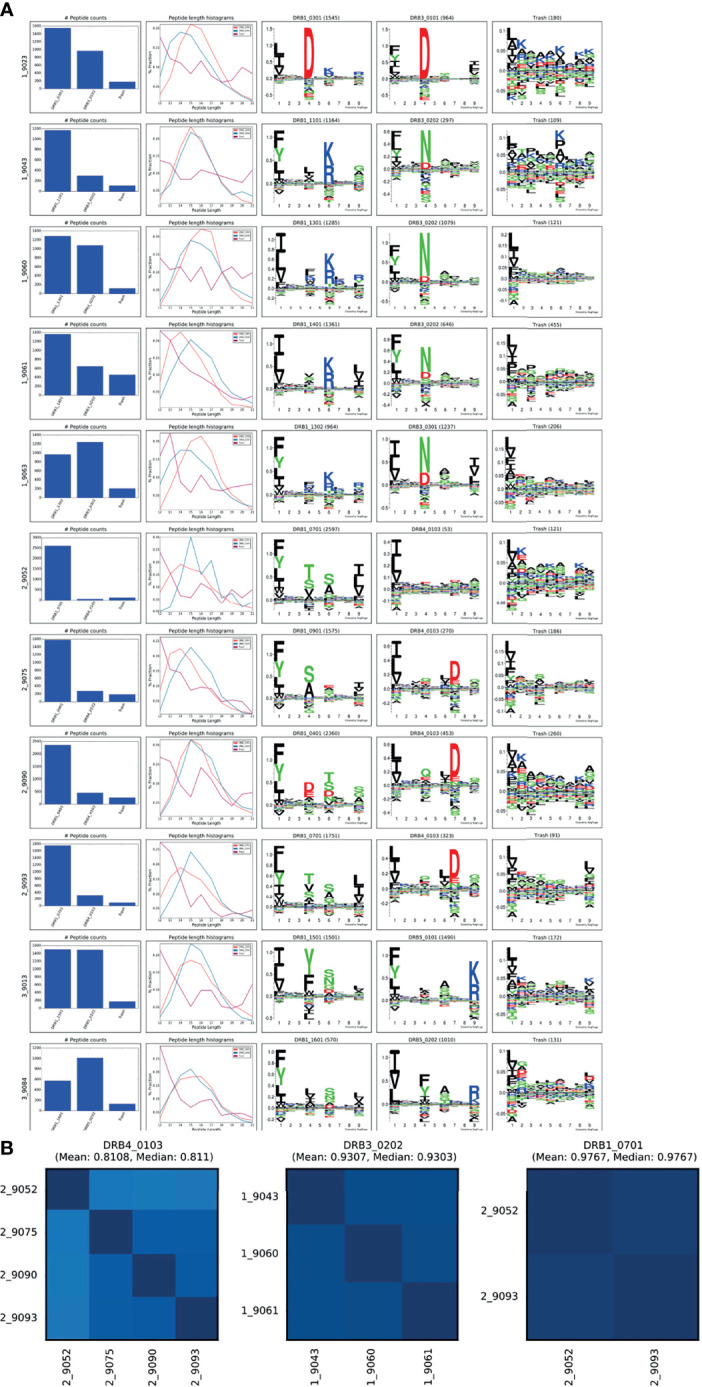
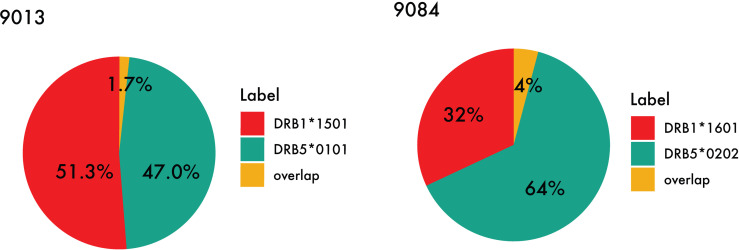
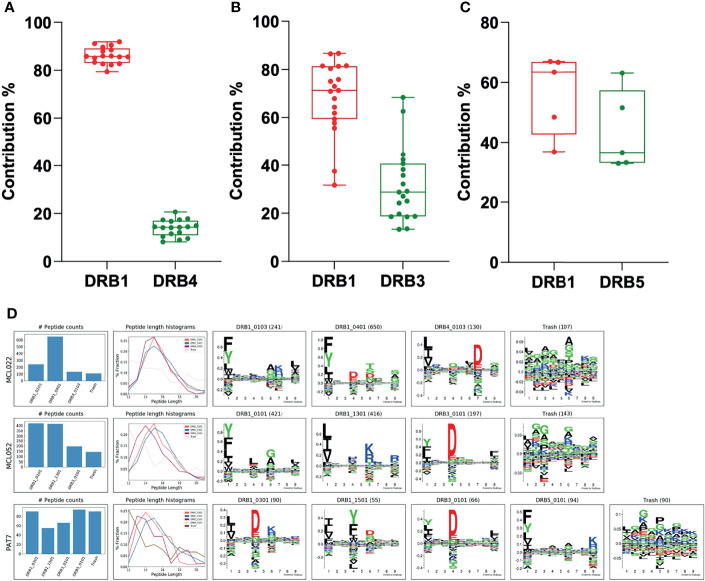
Mass spectrometry (MS) based immunopeptidomics is used in several biomedical applications including neo-epitope discovery in oncology, next-generation vaccine development and protein-drug immunogenicity assessment. Immunopeptidome data are highly complex given the expression of multiple HLA alleles on the cell membrane and presence of co-immunoprecipitated contaminants. The absence of tools that deal with these challenges effectively and guide the analysis and interpretation of this complex type of data is currently a major bottleneck for the large-scale application of this technique. To resolve this, we here present the MHCMotifDecon that benefits from state-of-the-art HLA class-I and class-II predictions to accurately deconvolute immunopeptidome datasets and assign individual ligands to the most likely HLA molecule, allowing to identify and characterize HLA binding motifs while discarding co-purified contaminants. We have benchmarked the tool against other state-of-the-art methods and illustrated its application on experimental datasets for HLA-DR demonstrating a previously underappreciated role for HLA-DRB3/4/5 molecules in defining HLA class II immune repertoires. With its ease of use, MHCMotifDecon can efficiently guide interpretation of immunopeptidome datasets, serving the discovery of novel T cell targets. MHCMotifDecon is available at https://services.healthtech.dtu.dk/service.php?MHCMotifDecon-1.0.
Keywords: DRB3/4/5; MHC motif deconvolution; MHCMotifDecon; immunopeptidome; mass spectrometry.
Copyright © 2022 Kaabinejadian, Barra, Alvarez, Yari, Hildebrand and Nielsen.
Conflict of interest statement
SK is an employee at Pure MHC, LLC. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
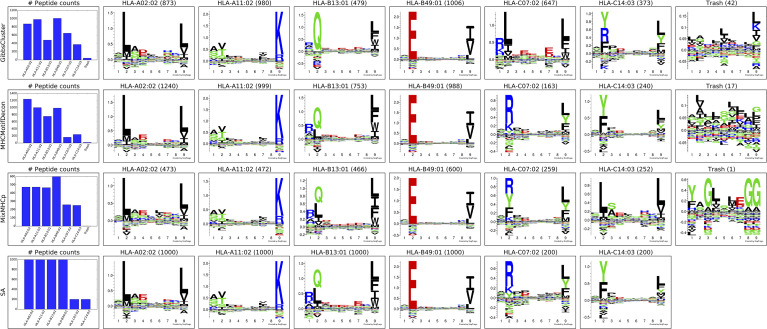
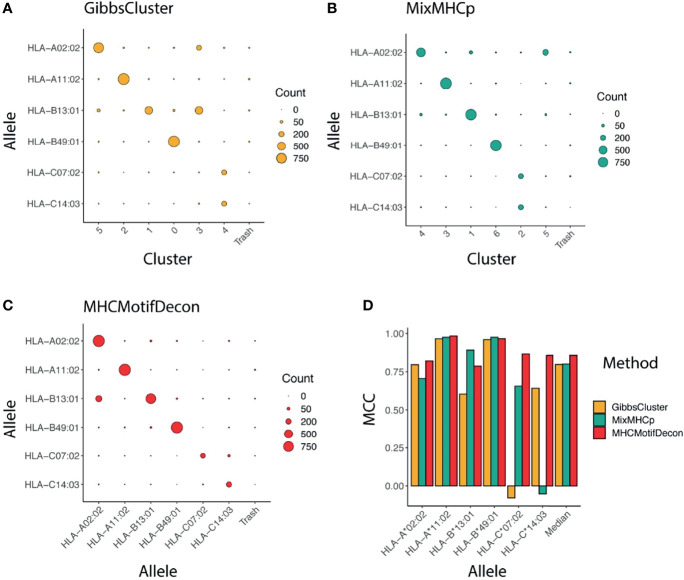

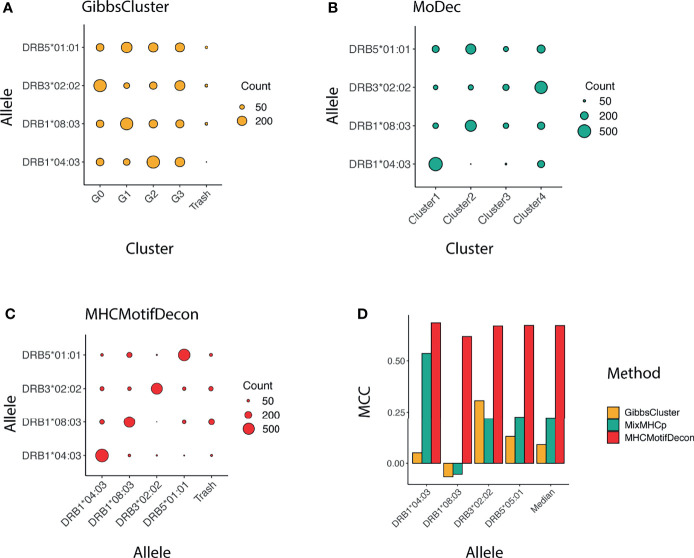
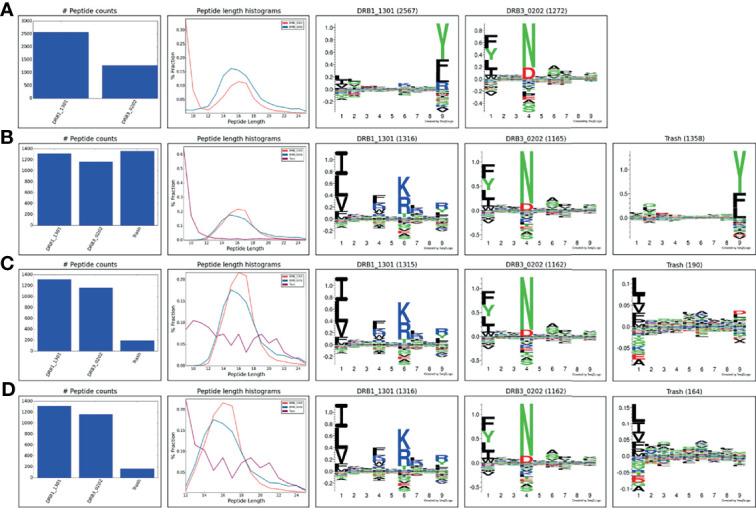
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials