

Dual Inhibition of Myc Transcription and PI3K Activity Effectively Targets Colorectal Cancer Stem Cells
- PMID: 35158939
- PMCID: PMC8833549
- DOI: 10.3390/cancers14030673
Dual Inhibition of Myc Transcription and PI3K Activity Effectively Targets Colorectal Cancer Stem Cells
Abstract
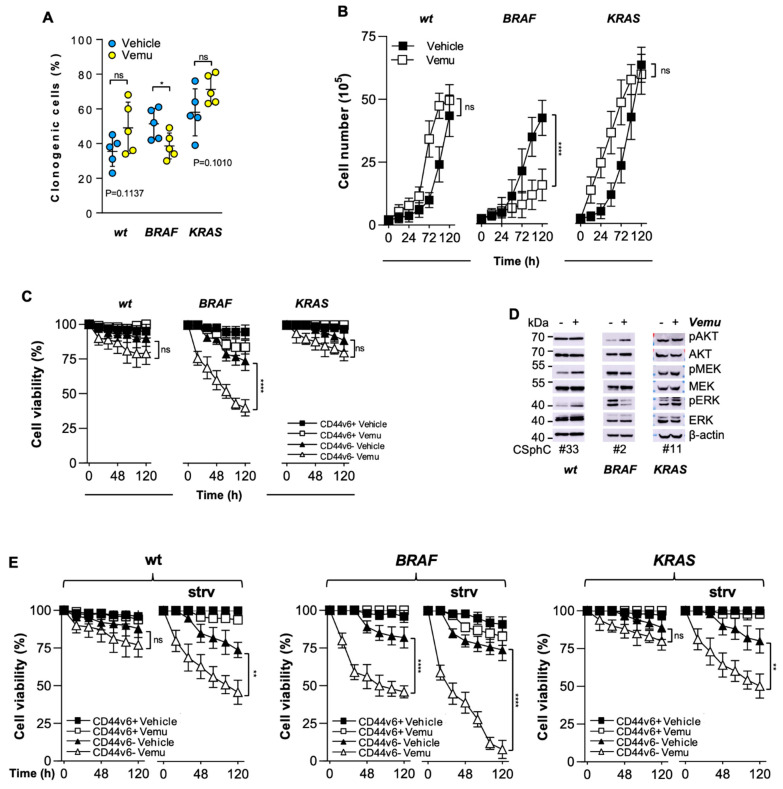
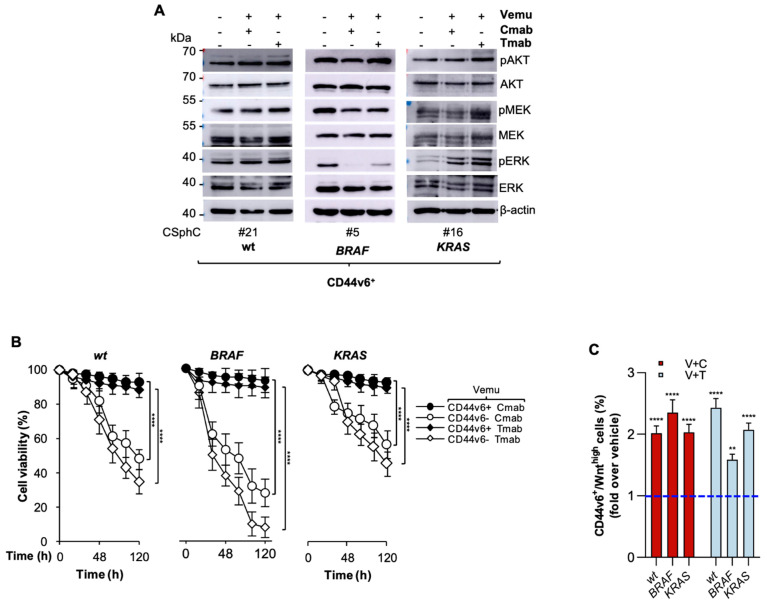
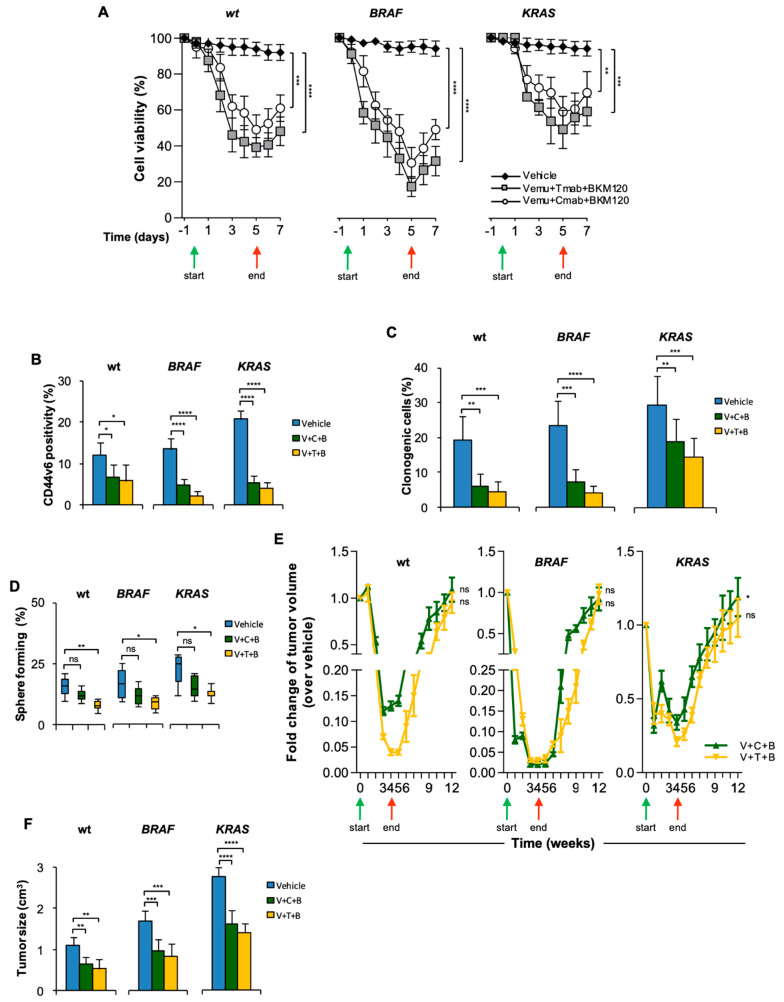
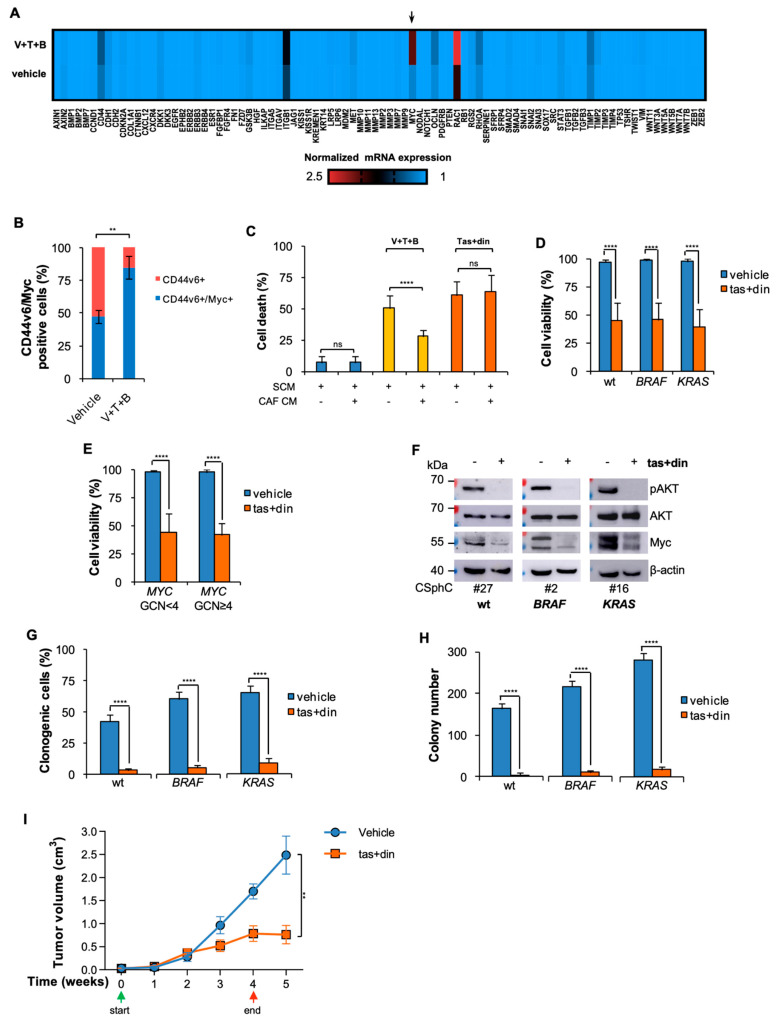
Despite advances in the curative approach, the survival rate of advanced colorectal cancer (CRC) patients is still poor, which is likely due to the emergence of cancer cell clones resistant to the available therapeutic options. We have already shown that CD44v6-positive CRC stem cells (CR-CSCs) are refractory toward standard anti-tumor therapeutic agents due to the activation of the PI3K pathway together with high HER2 expression levels. Tumor microenvironmental cytokines confer resistance to CR-CSCs against HER2/PI3K targeting by enhancing activation of the MAPK pathway. Here, we show that the CSC compartment, spared by BRAF inhibitor-based targeted therapy, is associated with increased expression levels of CD44v6 and Myc and retains boosted clonogenic activity along with residual tumorigenic potential. Inhibition of Myc transcription, downstream of the MAPK cascade components, and PI3K pathway activity was able to overcome the protective effects of microenvironmental cytokines, affecting the survival and the clonogenic activity of CR-CSCs, regardless of their mutational background. Likewise, the double targeting induced stabilization of mouse tumor avatars. Altogether, these data outline the rationale for dual kinase targeting of CR-CSCs to prevent their adaptive response, which would lead to disease progression.
Keywords: anti-tumor drug resistance; cancer stem cells; colorectal cancer; combination therapies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Gaggianesi M., Di Franco S., Pantina V.D., Porcelli G., D’Accardo C., Verona F., Veschi V., Colarossi L., Faldetta N., Pistone G., et al. Messing Up the Cancer Stem Cell Chemoresistance Mechanisms Supported by Tumor Microenvironment. Front. Oncol. 2021;11:2847. doi: 10.3389/fonc.2021.702642. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
