

Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI)
- PMID: 35159128
- PMCID: PMC8834229
- DOI: 10.3390/cells11030318
Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI)
Abstract
STING-associated vasculopathy with onset in infancy (SAVI) is a type I interferonopathy caused by gain-of-function mutations in STING1 encoding stimulator of interferon genes (STING) protein. SAVI is characterized by severe inflammatory lung disease, a feature not observed in previously described type I interferonopathies i.e., Mendelian autoinflammatory disorders defined by constitutive activation of the type I interferon (IFN) pathway. Molecular defects in nucleic acid metabolism or sensing are central to the pathophysiology of these diseases, with such defects occurring at any step of the tightly regulated pathway of type I IFN production and signaling (e.g., exonuclease loss of function, RNA-DNA hybrid accumulation, constitutive activation of adaptor proteins such as STING). Among over 30 genotypes, SAVI and COPA syndrome, whose pathophysiology was recently linked to a constitutive activation of STING signaling, are the only type I interferonopathies presenting with predominant lung involvement. Lung disease is the leading cause of morbidity and mortality in these two disorders which do not respond to conventional immunosuppressive therapies and only partially to JAK1/2 inhibitors. In human silicosis, STING-dependent sensing of self-DNA following cell death triggered by silica exposure has been found to drive lung inflammation in mice and human models. These recent findings support a key role for STING and nucleic acid sensing in the homeostasis of intrinsic pulmonary inflammation. However, mechanisms by which monogenic defects in the STING pathway lead to pulmonary damages are not yet fully elucidated, and an improved understanding of such mechanisms is fundamental to improved future patient management. Here, we review the recent insights into the pathophysiology of SAVI and outline our current understanding of self-nucleic acid-mediated lung inflammation in humans.
Keywords: STING-associated vasculopathy with onset in infancy; interferons; nucleic acid sensing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
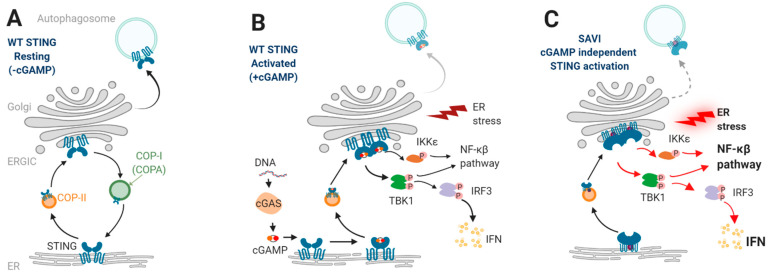
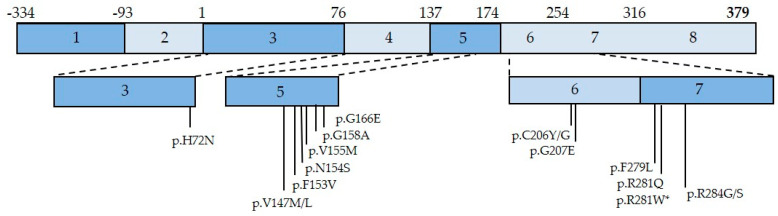
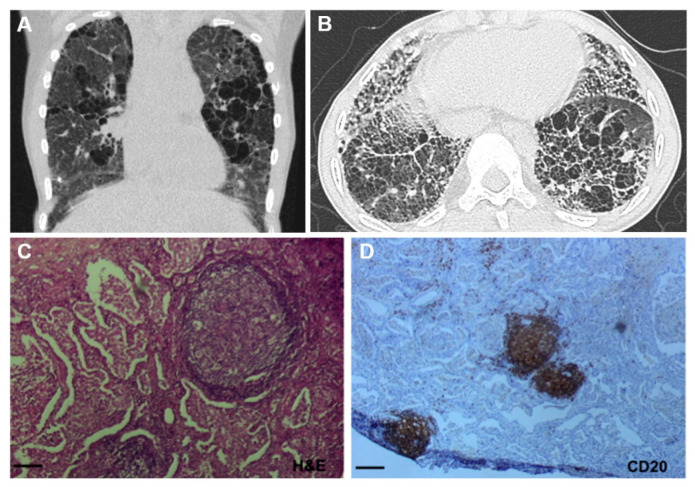
References
-
- Crow Y.J., Chase D.S., Lowenstein Schmidt J., Szynkiewicz M., Forte G.M.A., Gornall H.L., Oojageer A., Anderson B., Pizzino A., Helman G., et al. Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A. 2015;167A:296–312. doi: 10.1002/ajmg.a.36887. - DOI - PMC - PubMed
-
- Pokatayev V., Hasin N., Chon H., Cerritelli S.M., Sakhuja K., Ward J.M., Morris H.D., Yan N., Crouch R.J. RNase H2 Catalytic Core Aicardi-Goutières Syndrome-Related Mutant Invokes CGAS-STING Innate Immune-Sensing Pathway in Mice. J. Exp. Med. 2016;213:329–336. doi: 10.1084/jem.20151464. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
