

Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy
- PMID: 35159227
- PMCID: PMC8834523
- DOI: 10.3390/cells11030417
Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy
Abstract
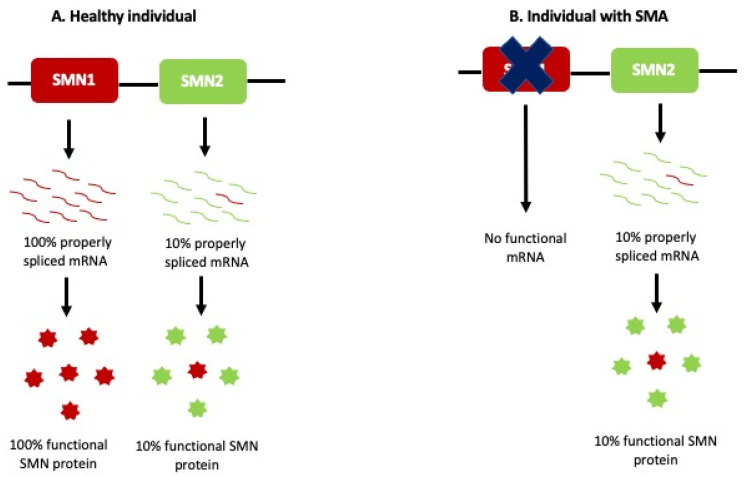
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder and one of the most common genetic causes of infant death. It is characterized by progressive weakness of the muscles, loss of ambulation, and death from respiratory complications. SMA is caused by the homozygous deletion or mutations in the survival of the motor neuron 1 (SMN1) gene. Humans, however, have a nearly identical copy of SMN1 known as the SMN2 gene. The severity of the disease correlates inversely with the number of SMN2 copies present. SMN2 cannot completely compensate for the loss of SMN1 in SMA patients because it can produce only a fraction of functional SMN protein. SMN protein is ubiquitously expressed in the body and has a variety of roles ranging from assembling the spliceosomal machinery, autophagy, RNA metabolism, signal transduction, cellular homeostasis, DNA repair, and recombination. Motor neurons in the anterior horn of the spinal cord are extremely susceptible to the loss of SMN protein, with the reason still being unclear. Due to the ability of the SMN2 gene to produce small amounts of functional SMN, two FDA-approved treatment strategies, including an antisense oligonucleotide (AON) nusinersen and small-molecule risdiplam, target SMN2 to produce more functional SMN. On the other hand, Onasemnogene abeparvovec (brand name Zolgensma) is an FDA-approved adeno-associated vector 9-mediated gene replacement therapy that can deliver a copy of the human SMN1. In this review, we summarize the SMA etiology, the role of SMN, and discuss the challenges of the therapies that are approved for SMA treatment.
Keywords: SMN protein; SMN2; antisense oligonucleotide (AON); gene therapy; nusinersen; onasemnogene; risdiplam; small molecule; spinal muscular atrophy (SMA); survival of motor neuron 1 (SMN1).
Conflict of interest statement
T.Y. is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
