

Epiregulin as an Alternative Ligand for Leptin Receptor Alleviates Glucose Intolerance without Change in Obesity
- PMID: 35159237
- PMCID: PMC8834548
- DOI: 10.3390/cells11030425
Epiregulin as an Alternative Ligand for Leptin Receptor Alleviates Glucose Intolerance without Change in Obesity
Abstract
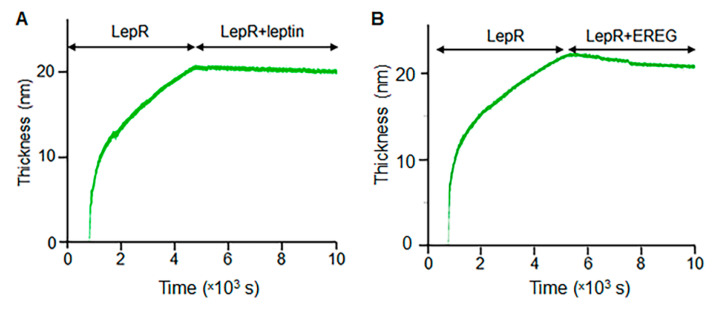
The leptin receptor (LepR) acts as a signaling nexus for the regulation of glucose uptake and obesity, among other metabolic responses. The functional role of LepR under leptin-deficient conditions remains unclear. This study reports that epiregulin (EREG) governed glucose uptake in vitro and in vivo in Lepob mice by activating LepR under leptin-deficient conditions. Single and long-term treatment with EREG effectively rescued glucose intolerance in comparative insulin and EREG tolerance tests in Lepob mice. The immunoprecipitation study revealed binding between EREG and LepR in adipose tissue of Lepob mice. EREG/LepR regulated glucose uptake without changes in obesity in Lepob mice via mechanisms, including ERK activation and translocation of GLUT4 to the cell surface. EREG-dependent glucose uptake was abolished in Leprdb mice which supports a key role of LepR in this process. In contrast, inhibition of the canonical epidermal growth factor receptor (EGFR) pathway implicated in other EREG responses, increased glucose uptake. Our data provide a basis for understanding glycemic responses of EREG that are dependent on LepR unlike functions mediated by EGFR, including leptin secretion, thermogenesis, pain, growth, and other responses. The computational analysis identified a conserved amino acid sequence, supporting an evolutionary role of EREG as an alternative LepR ligand.
Keywords: EGFR; ERK; energy metabolism; epiregulin; glucose uptake; leptin receptor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Fujikawa T., Berglund E.D., Patel V.R., Ramadori G., Vianna C.R., Vong L., Thorel F., Chera S., Herrera P.L., Lowell B.B., et al. Leptin Engages a Hypothalamic Neurocircuitry to Permit Survival in the Absence of Insulin. Cell Metab. 2013;18:431–444. doi: 10.1016/j.cmet.2013.08.004. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
