

Prion Protein: The Molecule of Many Forms and Faces
- PMID: 35163156
- PMCID: PMC8835406
- DOI: 10.3390/ijms23031232
Prion Protein: The Molecule of Many Forms and Faces
Abstract
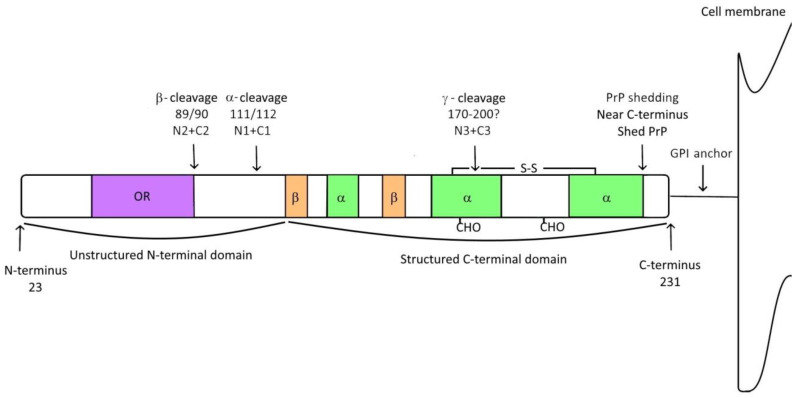
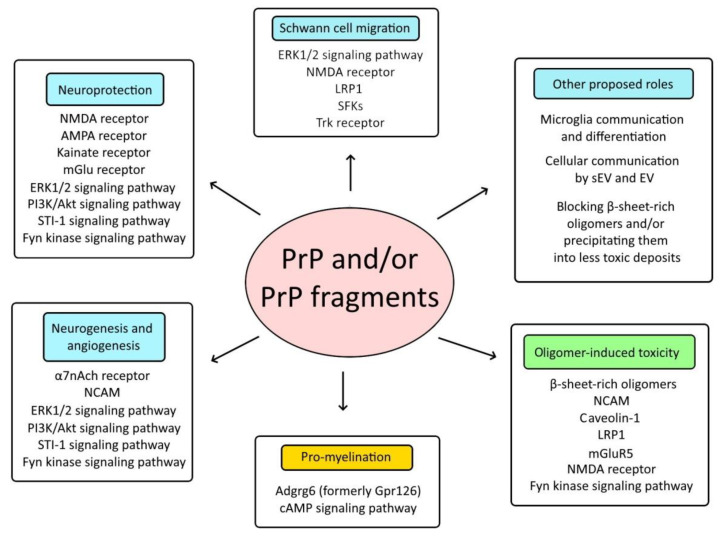
Cellular prion protein (PrPC) is a glycosylphosphatidylinositol (GPI)-anchored protein most abundantly found in the outer membrane of neurons. Due to structural characteristics (a flexible tail and structured core), PrPC interacts with a wide range of partners. Although PrPC has been proposed to be involved in many physiological functions, only peripheral nerve myelination homeostasis has been confirmed as a bona fide function thus far. PrPC misfolding causes prion diseases and PrPC has been shown to mediate β-rich oligomer-induced neurotoxicity in Alzheimer's and Parkinson's disease as well as neuroprotection in ischemia. Upon proteolytic cleavage, PrPC is transformed into released and attached forms of PrP that can, depending on the contained structural characteristics of PrPC, display protective or toxic properties. In this review, we will outline prion protein and prion protein fragment properties as well as overview their involvement with interacting partners and signal pathways in myelination, neuroprotection and neurodegenerative diseases.
Keywords: ischemic stroke; myelination; neurodegenerative disease; neuroprotection; prion protein; prion protein fragments.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
