

Gaucher Disease Diagnosis Using Lyso-Gb1 on Dry Blood Spot Samples: Time to Change the Paradigm?
- PMID: 35163551
- PMCID: PMC8835963
- DOI: 10.3390/ijms23031627
Gaucher Disease Diagnosis Using Lyso-Gb1 on Dry Blood Spot Samples: Time to Change the Paradigm?
Abstract
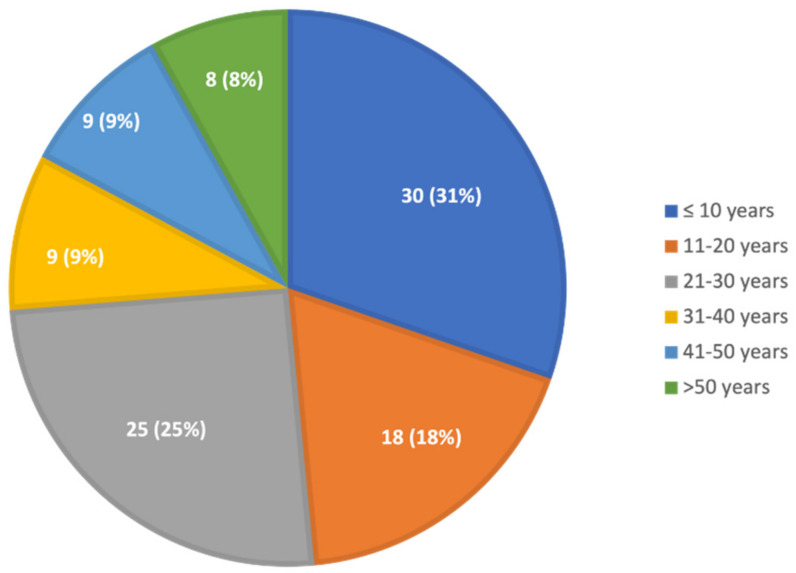
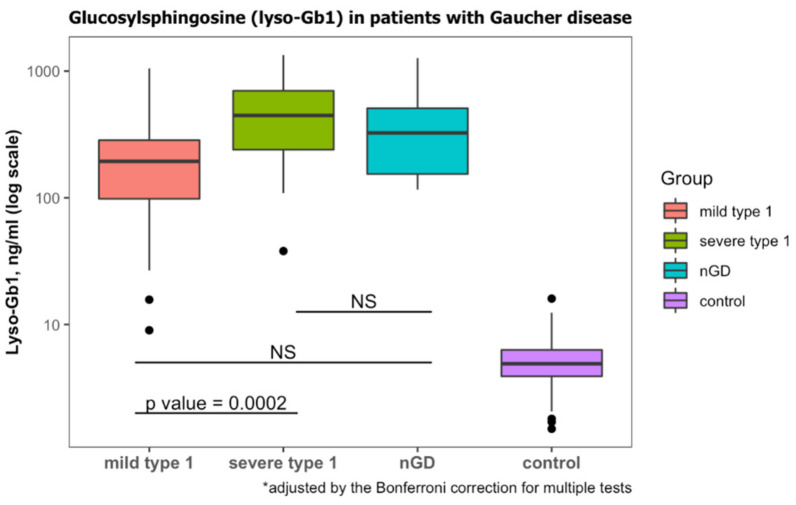
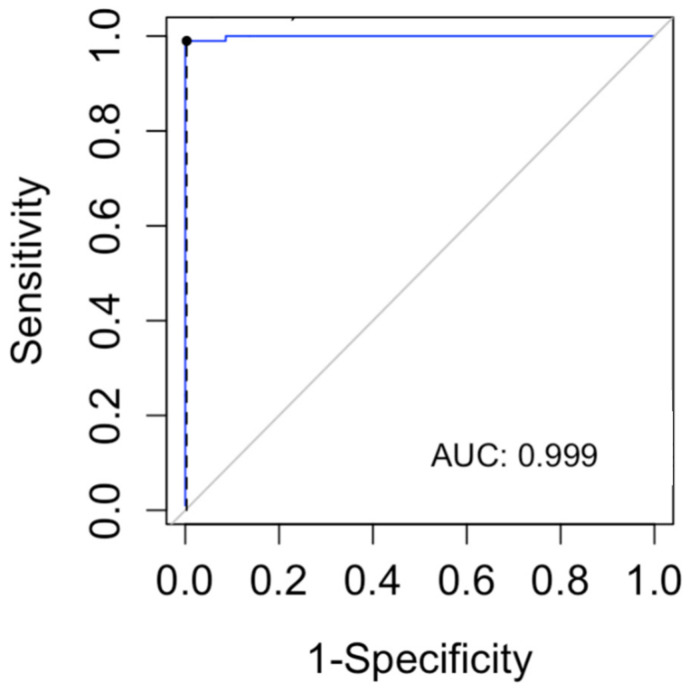
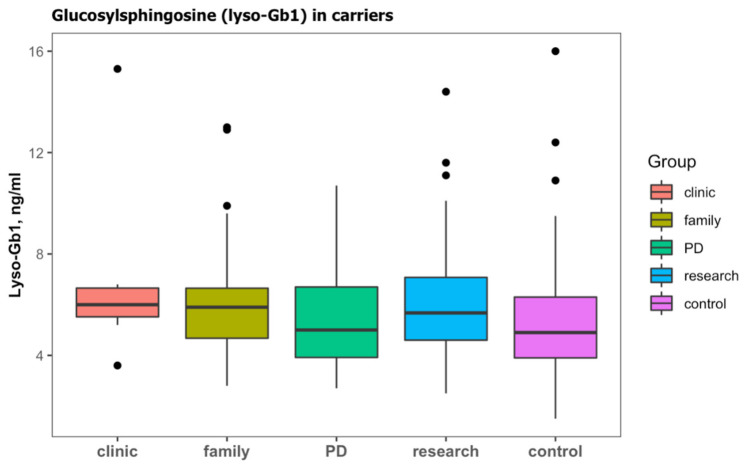
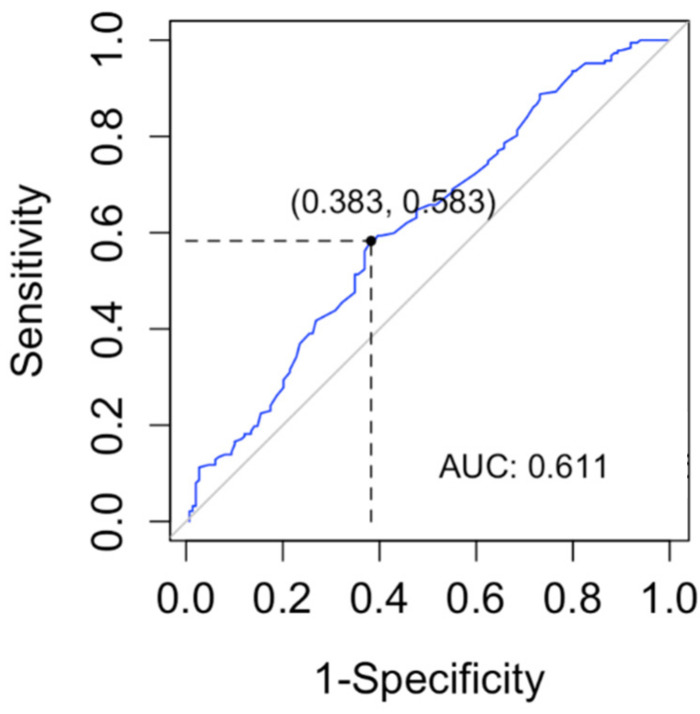
For years, the gold standard for diagnosing Gaucher disease (GD) has been detecting reduced β-glucocerebrosidase (GCase) activity in peripheral blood cells combined with GBA1 mutation analysis. The use of dried blood spot (DBS) specimens offers many advantages, including easy collection, the need for a small amount of blood, and simpler transportation. However, DBS has limitations for measuring GCase activity. In this paper, we recount our cross-sectional study and publish seven years of experience using DBS samples and levels of the deacylated form of glucocerebroside, glucosylsphingosine (lyso-Gb1), for GD diagnosis. Of 444 screened subjects, 99 (22.3%) were diagnosed with GD at a median (range) age of 21 (1-78) years. Lyso-Gb levels for genetically confirmed GD patients vs. subjects negative to GD diagnosis were 252 (9-1340) ng/mL and 5.4 (1.5-16) ng/mL, respectively. Patients diagnosed with GD1 and mild GBA1 variants had lower median (range) lyso-Gb1, 194 (9-1050), compared to GD1 and severe GBA1 variants, 447 (38-1340) ng/mL, and neuronopathic GD, 325 (116-1270) ng/mL (p = 0.001). Subjects with heterozygous GBA1 variants (carrier) had higher lyso-Gb1 levels, 5.8 (2.5-15.3) ng/mL, compared to wild-type GBA1, 4.9 (1.5-16), ng/mL (p = 0.001). Lyso-Gb1 levels, median (range), were 5 (2.7-10.7) in heterozygous GBA1 carriers with Parkinson's disease (PD), similar to lyso-Gb1 levels in subjects without PD. We call for a paradigm change for the diagnosis of GD based on lyso-Gb1 measurements and confirmatory GBA1 mutation analyses in DBS. Lyso-Gb1 levels could not be used to differentiate between heterozygous GBA1 carriers and wild type.
Keywords: Gaucher disease; diagnosis; dry blood spot; glucosylsphingosine; lyso-Gb1.
Conflict of interest statement
The SZMC Gaucher Unit receives support from Sanofi/Genzyme for participation in the ICGG Registry, from Takeda for the GOS Registry, and Pfizer for TALIAS. The Unit also receives research grants from Takeda, Pfizer, Sanofi/Genzyme, and Centogene. T.D., M.B.-C. and M.I. have no conflict of interest to declare. P.B., C.B., G.K., C.C. and M-I.I. are employees of Centogene GmbH. A.R. is the founder and was the CEO of Centogene GmbH during the study. A.Z. receives honoraria from Takeda, Pfizer, and BioEvents and consultancy fees from Takeda, NLC Pharma, Insightec, and Prevail therapeutics. S.R.-V. receives grant/research support, honoraria, and advisory fee from Takeda, Pfizer, and Sanofi/Genzyme.
Figures
Similar articles
-
Detection of glucosylsphingosine in dried blood spots for diagnosis of Gaucher disease by LC-MS/MS.Clin Biochem. 2021 Jan;87:79-84. doi: 10.1016/j.clinbiochem.2020.10.011. Epub 2020 Nov 11. Clin Biochem. 2021. PMID: 33188770
-
Effects of GBA1 Variants and Prenatal Exposition on the Glucosylsphingosine (Lyso-Gb1) Levels in Gaucher Disease Carriers.Int J Mol Sci. 2024 Nov 8;25(22):12021. doi: 10.3390/ijms252212021. Int J Mol Sci. 2024. PMID: 39596090 Free PMC article.
-
Contribution of Glucosylsphingosine (Lyso-Gb1) to Treatment Decisions in Patients with Gaucher Disease.Int J Mol Sci. 2023 Feb 15;24(4):3945. doi: 10.3390/ijms24043945. Int J Mol Sci. 2023. PMID: 36835356 Free PMC article.
-
Glucosylsphingosine (Lyso-Gb1) as a reliable biomarker in Gaucher disease: a narrative review.Orphanet J Rare Dis. 2023 Feb 13;18(1):27. doi: 10.1186/s13023-023-02623-7. Orphanet J Rare Dis. 2023. PMID: 36782327 Free PMC article. Review.
-
Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review.Int J Mol Sci. 2020 Sep 28;21(19):7159. doi: 10.3390/ijms21197159. Int J Mol Sci. 2020. PMID: 32998334 Free PMC article.
Cited by
-
Patient centered guidelines for the laboratory diagnosis of Gaucher disease type 1.Orphanet J Rare Dis. 2022 Dec 21;17(1):442. doi: 10.1186/s13023-022-02573-6. Orphanet J Rare Dis. 2022. PMID: 36544230 Free PMC article.
-
Real-Life Experience with Oral Eliglustat in Patients with Gaucher Disease Previously Treated with Enzyme Replacement Therapy.J Clin Med. 2022 Oct 24;11(21):6265. doi: 10.3390/jcm11216265. J Clin Med. 2022. PMID: 36362492 Free PMC article.
-
Diagnosis and genetic analysis of Gaucher disease in a pediatric case: a case report.Front Pediatr. 2025 Jul 28;13:1628525. doi: 10.3389/fped.2025.1628525. eCollection 2025. Front Pediatr. 2025. PMID: 40791806 Free PMC article.
-
High-Dose Ambroxol Therapy in Type 1 Gaucher Disease Focusing on Patients with Poor Response to Enzyme Replacement Therapy or Substrate Reduction Therapy.Int J Mol Sci. 2023 Apr 4;24(7):6732. doi: 10.3390/ijms24076732. Int J Mol Sci. 2023. PMID: 37047707 Free PMC article.
-
Long- and Short-Term Glucosphingosine (lyso-Gb1) Dynamics in Gaucher Patients Undergoing Enzyme Replacement Therapy.Biomolecules. 2024 Jul 12;14(7):842. doi: 10.3390/biom14070842. Biomolecules. 2024. PMID: 39062556 Free PMC article.
References
-
- Revel-Vilk S., Szer J., Zimran A. Gaucher disease and related lysosomal storage diseases. In: Kaushansky K., Lichtman M., Prchal J., Levi M., Press O., Burns L., Caligiuri M., editors. Williams Hematology. 10th ed. McGraw-Hill; New York, NY, USA: 2021. pp. 1189–1202.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
