

Conformational Flexibility and Local Frustration in the Functional States of the SARS-CoV-2 Spike B.1.1.7 and B.1.351 Variants: Mutation-Induced Allosteric Modulation Mechanism of Functional Dynamics and Protein Stability
- PMID: 35163572
- PMCID: PMC8836237
- DOI: 10.3390/ijms23031646
Conformational Flexibility and Local Frustration in the Functional States of the SARS-CoV-2 Spike B.1.1.7 and B.1.351 Variants: Mutation-Induced Allosteric Modulation Mechanism of Functional Dynamics and Protein Stability
Abstract
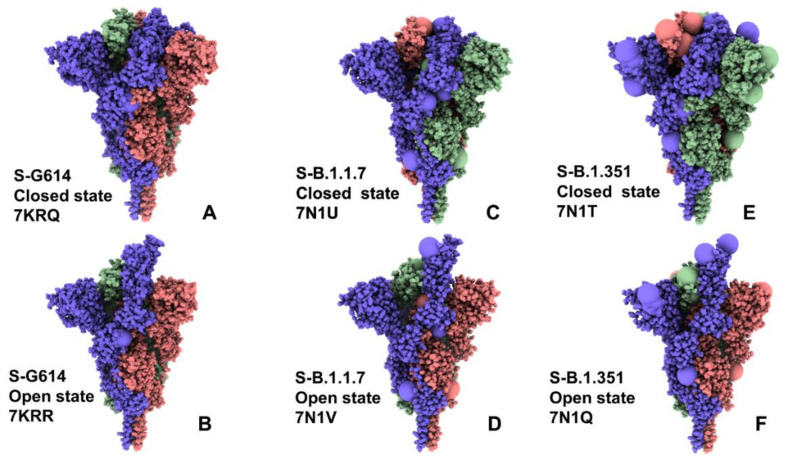
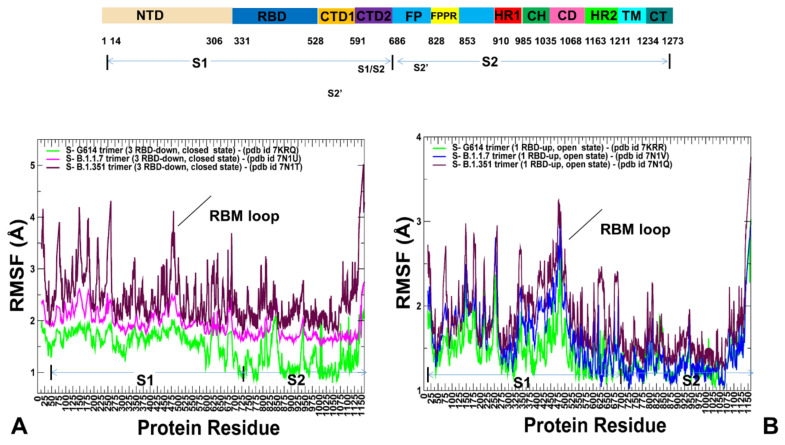
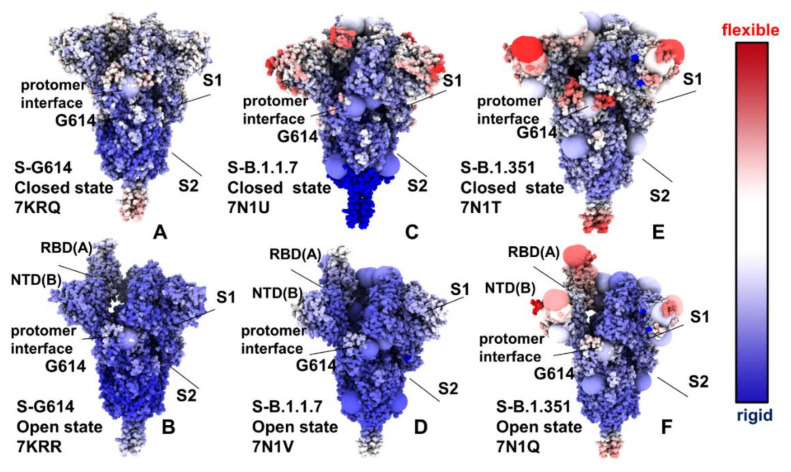
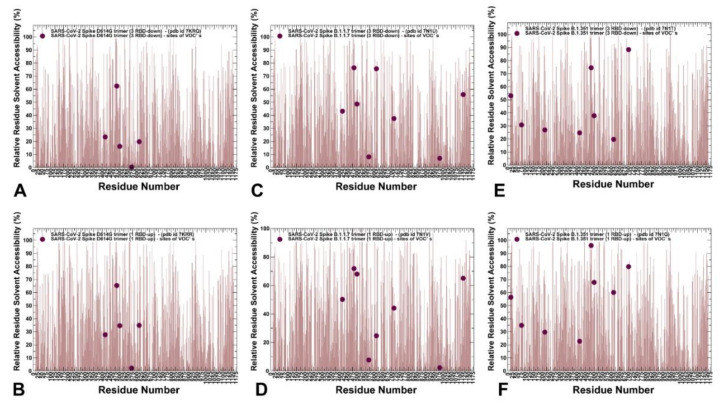
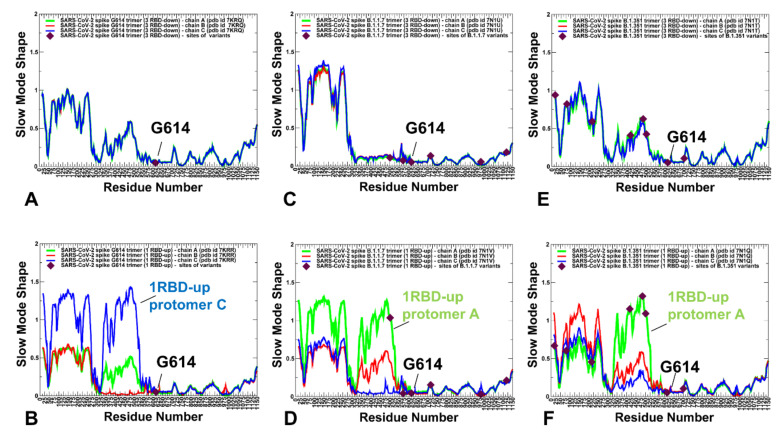
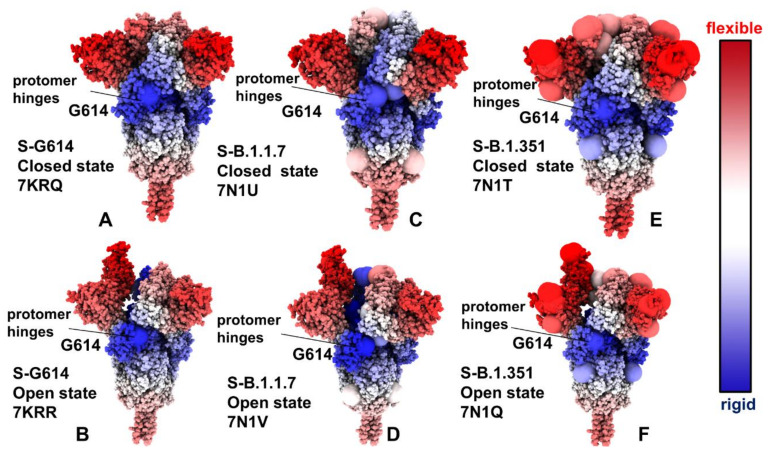
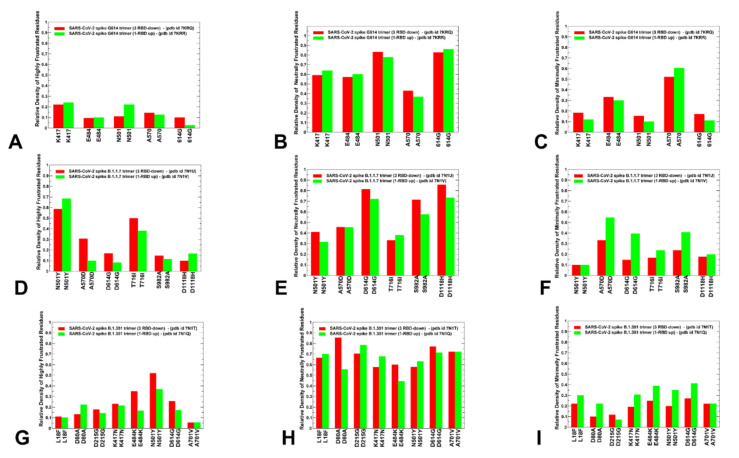
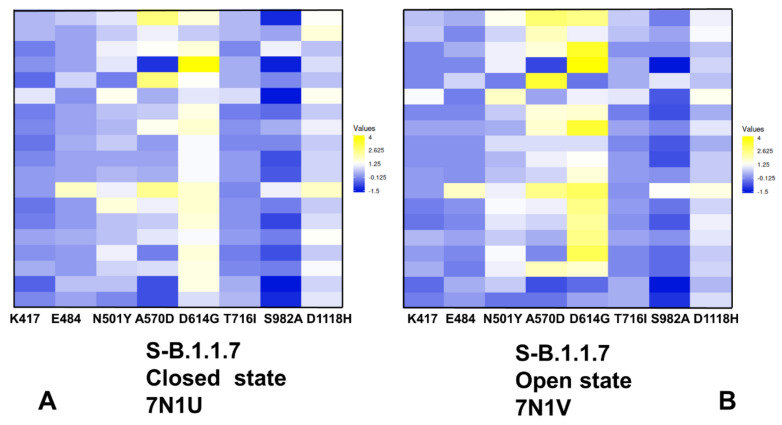
Structural and functional studies of the SARS-CoV-2 spike proteins have recently determined distinct functional states of the B.1.1.7 and B.1.351 spike variants, providing a molecular framework for understanding the mechanisms that link the effect of mutations with the enhanced virus infectivity and transmissibility. A detailed dynamic and energetic analysis of these variants was undertaken in the present work to quantify the effects of different mutations on functional conformational changes and stability of the SARS-CoV-2 spike protein. We employed the efficient and accurate coarse-grained (CG) simulations of multiple functional states of the D614G mutant, B.1.1.7 and B.1.351 spike variants to characterize conformational dynamics of the SARS-CoV-2 spike proteins and identify dynamic signatures of the functional regions that regulate transitions between the closed and open forms. By combining molecular simulations with full atomistic reconstruction of the trajectories and the ensemble-based mutational frustration analysis, we characterized how the intrinsic flexibility of specific spike regions can control functional conformational changes required for binding with the host-cell receptor. Using the residue-based mutational scanning of protein stability, we determined protein stability hotspots and identified potential energetic drivers favoring the receptor-accessible open spike states for the B.1.1.7 and B.1.351 spike variants. The results suggested that modulation of the energetic frustration at the inter-protomer interfaces can serve as a mechanism for allosteric couplings between mutational sites and the inter-protomer hinges of functional motions. The proposed mechanism of mutation-induced energetic frustration may result in greater adaptability and the emergence of multiple conformational states in the open form. This study suggested that SARS-CoV-2 B.1.1.7 and B.1.351 variants may leverage the intrinsic plasticity of functional regions in the spike protein for mutation-induced modulation of protein dynamics and allosteric regulation to control binding with the host cell receptor.
Keywords: ACE2 host receptor; SARS-CoV-2 spike protein; allosteric interactions; conformational dynamics; local frustration; mutational scanning; mutational variants; protein stability.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
References
-
- Tai W., He L., Zhang X., Pu J., Voronin D., Jiang S., Zhou Y., Du L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020;17:613–620. doi: 10.1038/s41423-020-0400-4. - DOI - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
