

EphrinB2-EphB4 Signaling in Neurooncological Disease
- PMID: 35163601
- PMCID: PMC8836162
- DOI: 10.3390/ijms23031679
EphrinB2-EphB4 Signaling in Neurooncological Disease
Abstract
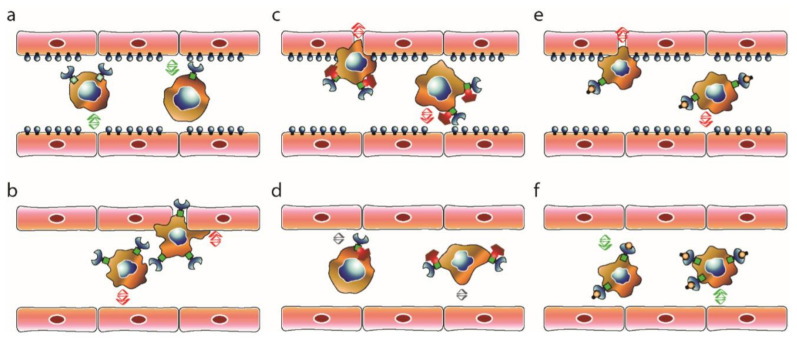
EphrinB2-EphB4 signaling is critical during embryogenesis for cardiovascular formation and neuronal guidance. Intriguingly, critical expression patterns have been discovered in cancer pathologies over the last two decades. Multiple connections to tumor migration, growth, angiogenesis, apoptosis, and metastasis have been identified in vitro and in vivo. However, the molecular signaling pathways are manifold and signaling of the EphB4 receptor or the ephrinB2 ligand is cancer type specific. Here we explore the impact of these signaling pathways in neurooncological disease, including glioma, brain metastasis, and spinal bone metastasis. We identify potential downstream pathways that mediate cancer suppression or progression and seek to understand it´s role in antiangiogenic therapy resistance in glioma. Despite the Janus-faced functions of ephrinB2-EphB4 signaling in cancer Eph signaling remains a promising clinical target.
Keywords: EphB4; bone; bone metastasis; brain metastasis; ephrinB2; glioblastoma; glioma; metastasis; neurooncology; spinal metastasis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
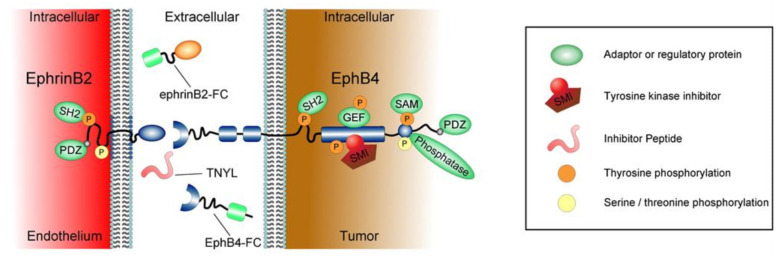
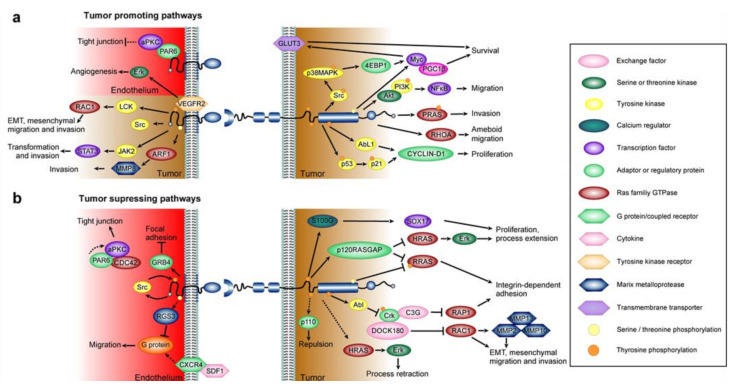
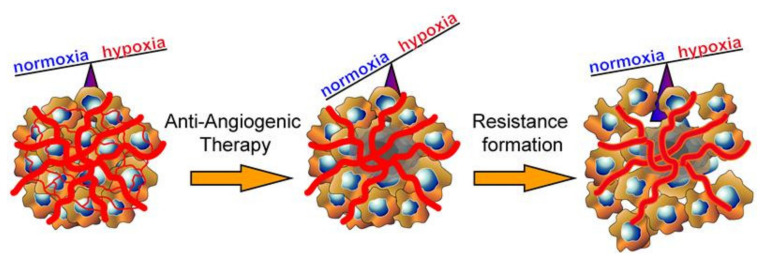
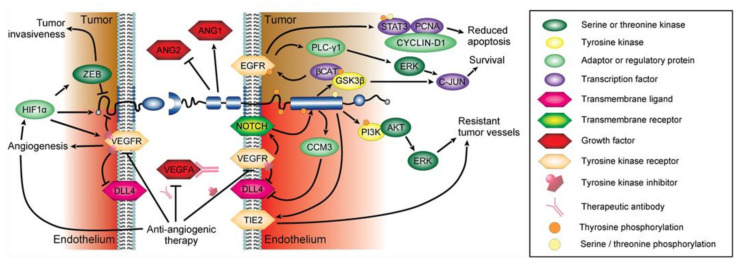
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
