

Hereditary Spastic Paraplegia: An Update
- PMID: 35163618
- PMCID: PMC8835766
- DOI: 10.3390/ijms23031697
Hereditary Spastic Paraplegia: An Update
Abstract
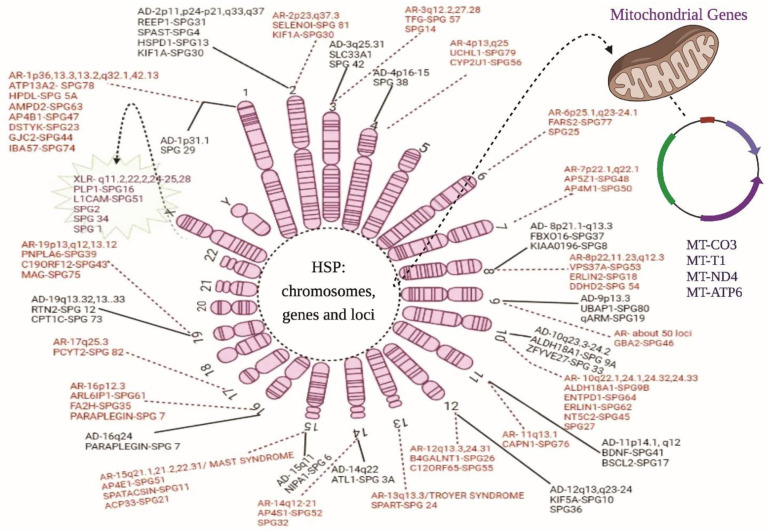
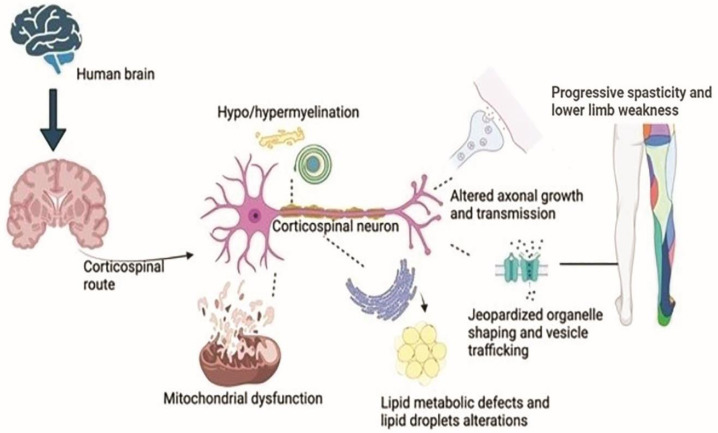
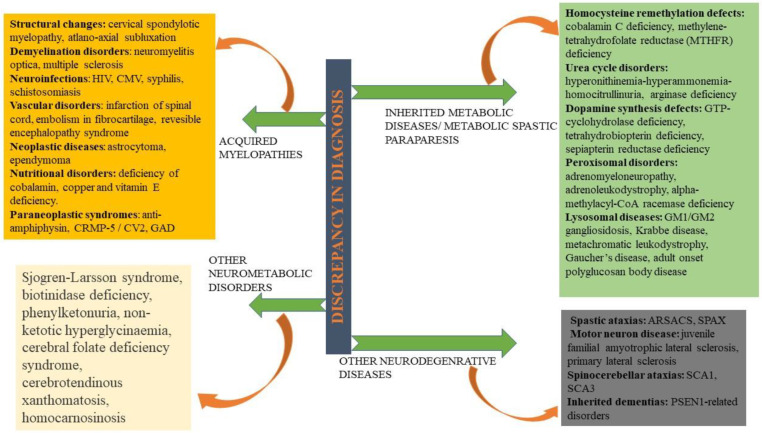
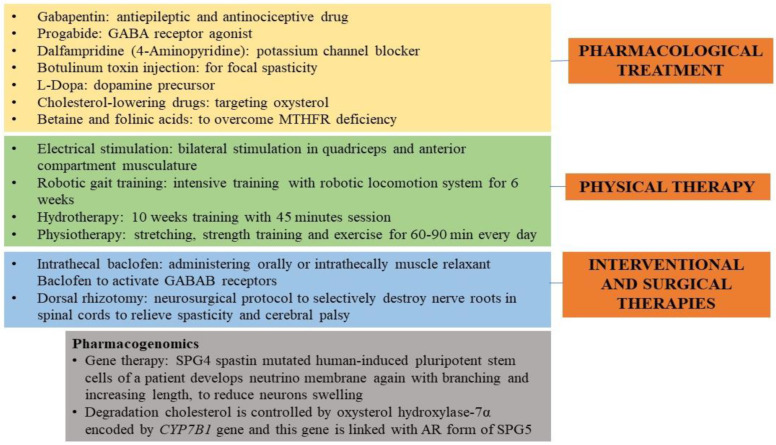
Hereditary spastic paraplegia (HSP) is a rare neurodegenerative disorder with the predominant clinical manifestation of spasticity in the lower extremities. HSP is categorised based on inheritance, the phenotypic characters, and the mode of molecular pathophysiology, with frequent degeneration in the axon of cervical and thoracic spinal cord's lateral region, comprising the corticospinal routes. The prevalence ranges from 0.1 to 9.6 subjects per 100,000 reported around the globe. Though modern medical interventions help recognize and manage the disorder, the symptomatic measures remain below satisfaction. The present review assimilates the available data on HSP and lists down the chromosomes involved in its pathophysiology and the mutations observed in the respective genes on the chromosomes. It also sheds light on the treatment available along with the oral/intrathecal medications, physical therapies, and surgical interventions. Finally, we have discussed the related diagnostic techniques as well as the linked pharmacogenomics studies under future perspectives.
Keywords: hereditary spastic paraplegia; neurodegenerative disease; neurogenetics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- De Beukelaer N., Bar-On L., Hanssen B., Peeters N., Prinsen S., Ortibus E., Desloovere K., Van Campenhout A. Muscle characteristics in pediatric hereditary spastic paraplegia vs. bilateral spastic cerebral palsy: An exploratory study. Front. Neurol. 2021;12:635032. doi: 10.3389/fneur.2021.635032. - DOI - PMC - PubMed
-
- Carosi L., Giudice T.L., Lullo M., Di Lombardi F., Babalini C., Gaudiello F., Marfia G.A., Massa R., Kawarai T., Orlacchio A. Hereditary spastic paraplegia:A novel mutation and expansion of the phenotype variability in SPG10. J. Neurol. Neurosurg. Psychiatry. 2015;86:702–704. doi: 10.1136/jnnp-2014-308625. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
