

CRISPR Therapeutics for Duchenne Muscular Dystrophy
- PMID: 35163754
- PMCID: PMC8836469
- DOI: 10.3390/ijms23031832
CRISPR Therapeutics for Duchenne Muscular Dystrophy
Abstract
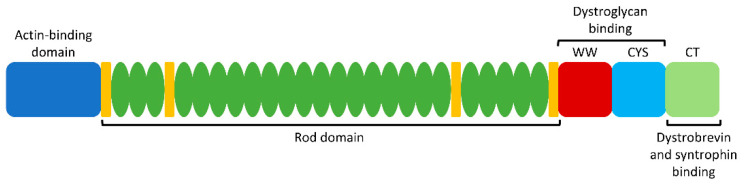
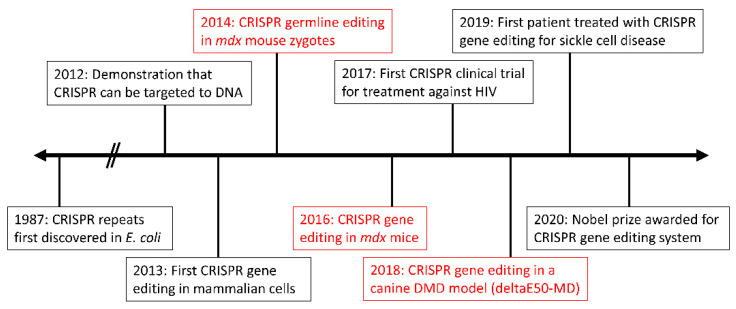
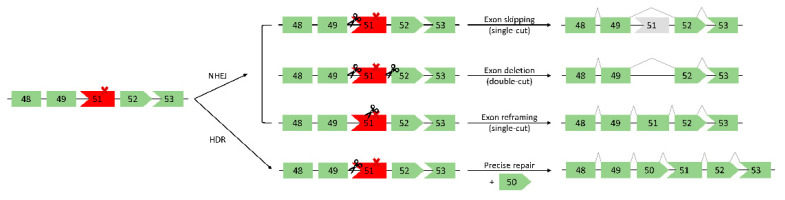
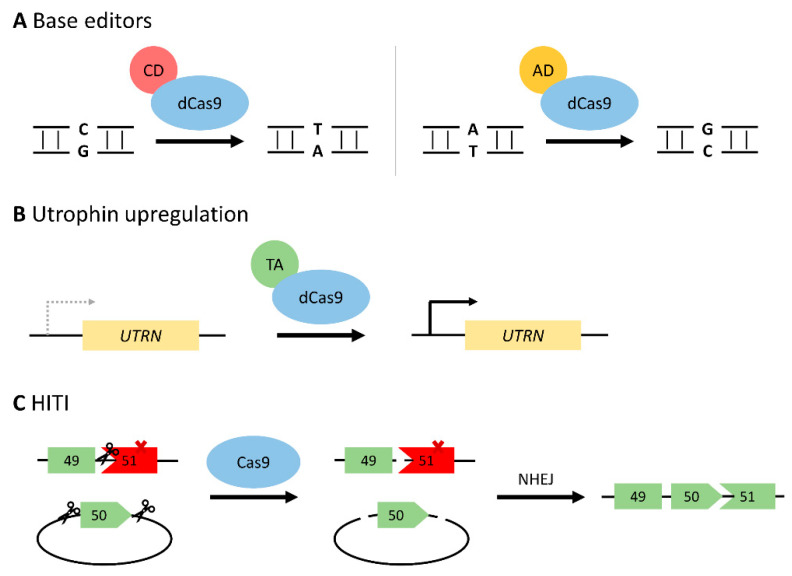
Duchenne muscular dystrophy (DMD) is an X-linked recessive neuromuscular disorder with a prevalence of approximately 1 in 3500-5000 males. DMD manifests as childhood-onset muscle degeneration, followed by loss of ambulation, cardiomyopathy, and death in early adulthood due to a lack of functional dystrophin protein. Out-of-frame mutations in the dystrophin gene are the most common underlying cause of DMD. Gene editing via the clustered regularly interspaced short palindromic repeats (CRISPR) system is a promising therapeutic for DMD, as it can permanently correct DMD mutations and thus restore the reading frame, allowing for the production of functional dystrophin. The specific mechanism of gene editing can vary based on a variety of factors such as the number of cuts generated by CRISPR, the presence of an exogenous DNA template, or the current cell cycle stage. CRISPR-mediated gene editing for DMD has been tested both in vitro and in vivo, with many of these studies discussed herein. Additionally, novel modifications to the CRISPR system such as base or prime editors allow for more precise gene editing. Despite recent advances, limitations remain including delivery efficiency, off-target mutagenesis, and long-term maintenance of dystrophin. Further studies focusing on safety and accuracy of the CRISPR system are necessary prior to clinical translation.
Keywords: CRISPR; Duchenne muscular dystrophy (DMD); NHEJ; dystrophin; exon skipping; gene editing.
Conflict of interest statement
TY is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology.
Figures
References
-
- Darras B.T., Menache-Starobinski C.C., Hinton V., Kunkel L.M. Chapter 30—Dystrophinopathies. In: Darras B.T., Jones H.R., Ryan M.M., De Vivo D.C., editors. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. 2nd ed. Academic Press; San Diego, CA, USA: 2015. pp. 551–592.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
