

Metabolic Features of Brain Function with Relevance to Clinical Features of Alzheimer and Parkinson Diseases
- PMID: 35164216
- PMCID: PMC8839962
- DOI: 10.3390/molecules27030951
Metabolic Features of Brain Function with Relevance to Clinical Features of Alzheimer and Parkinson Diseases
Abstract
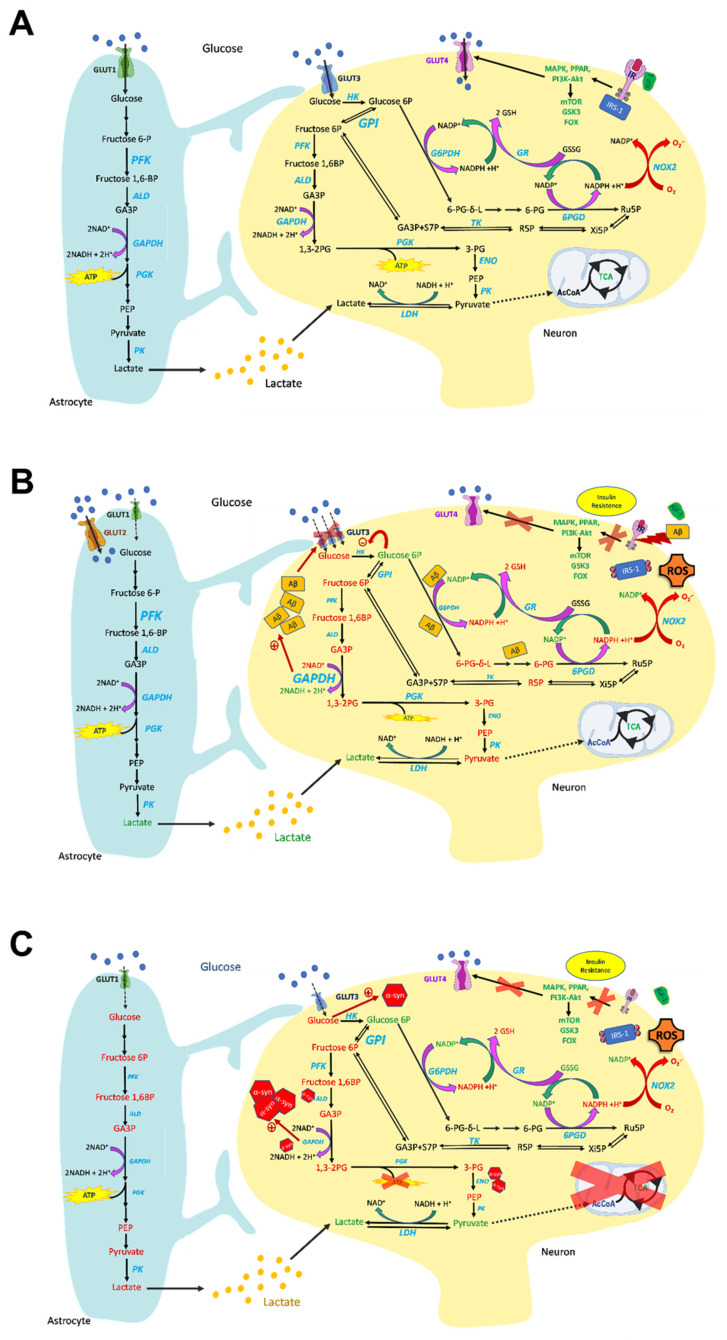
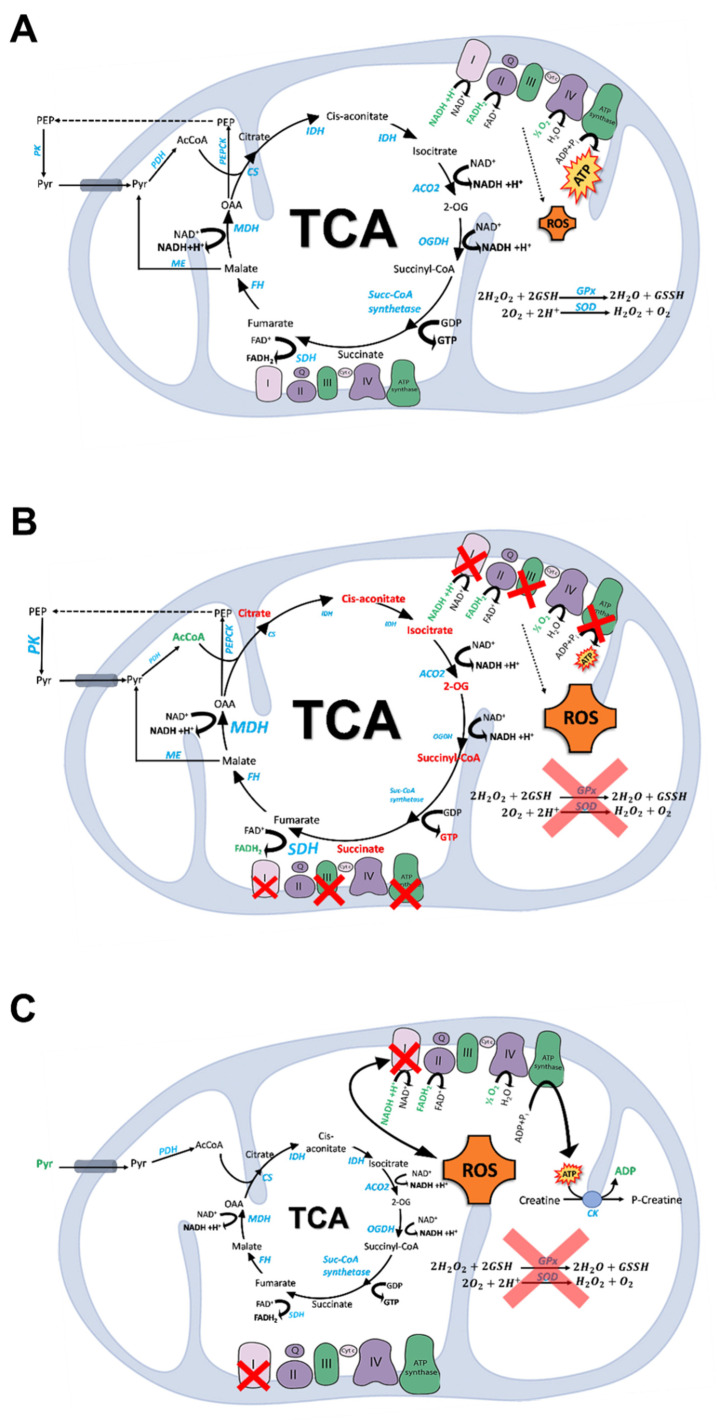
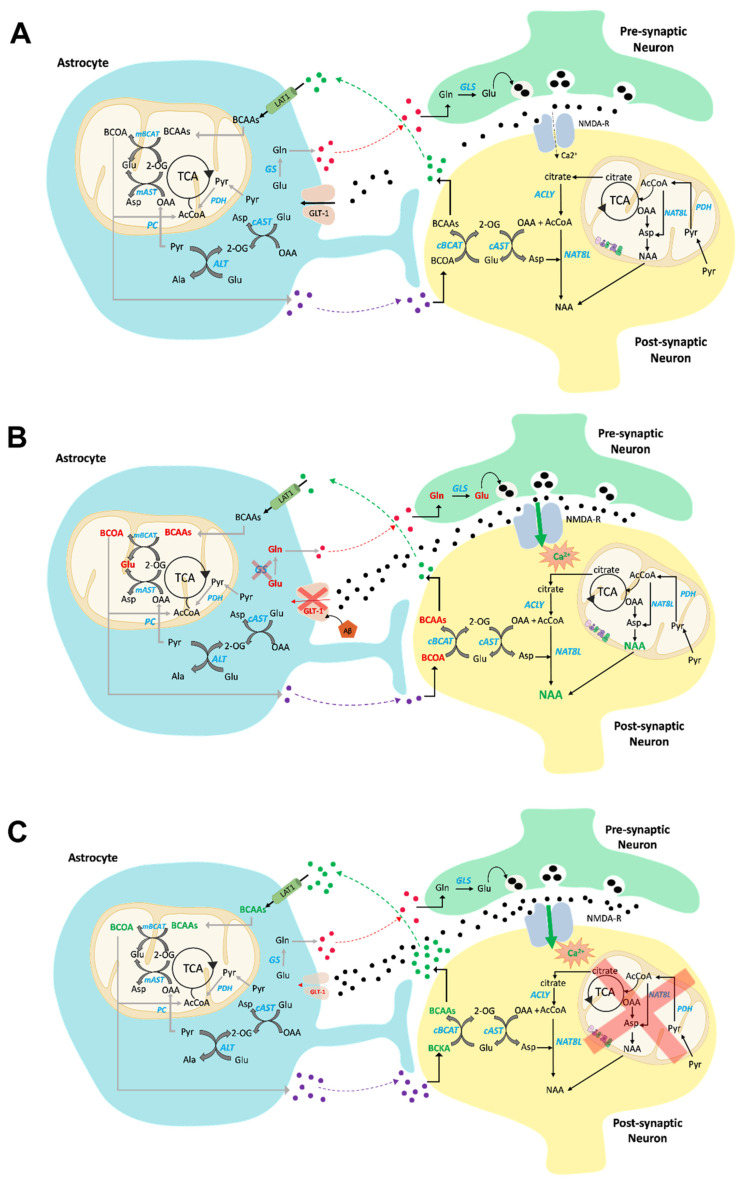
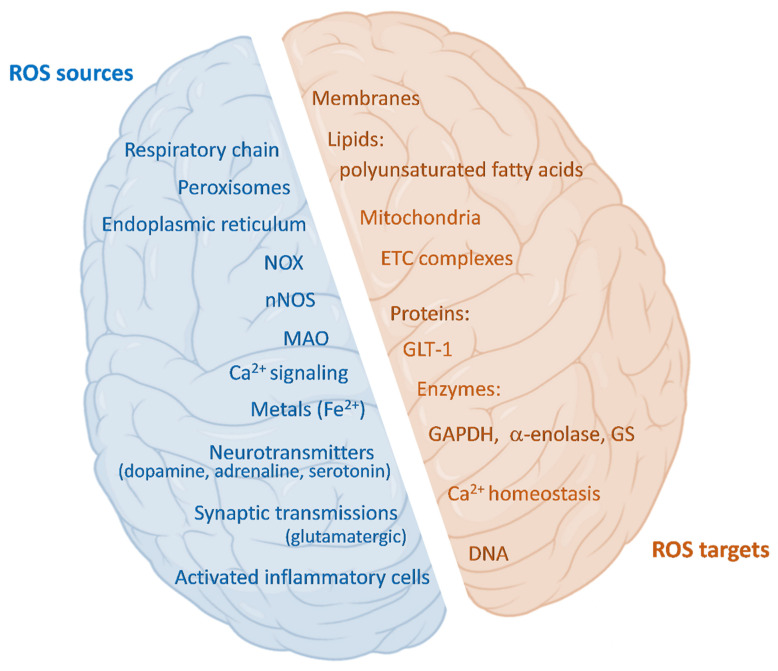
Brain metabolism is comprised in Alzheimer's disease (AD) and Parkinson's disease (PD). Since the brain primarily relies on metabolism of glucose, ketone bodies, and amino acids, aspects of these metabolic processes in these disorders-and particularly how these altered metabolic processes are related to oxidative and/or nitrosative stress and the resulting damaged targets-are reviewed in this paper. Greater understanding of the decreased functions in brain metabolism in AD and PD is posited to lead to potentially important therapeutic strategies to address both of these disorders, which cause relatively long-lasting decreased quality of life in patients.
Keywords: AD; Alzheimer’s disease; PD brain metabolism; Parkinson’s disease; glucose; metabolic reprogramming; neurodegeneration; oxidative stress.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases.Ann N Y Acad Sci. 1999;893:154-75. doi: 10.1111/j.1749-6632.1999.tb07824.x. Ann N Y Acad Sci. 1999. PMID: 10672236 Review.
-
Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases.Int J Mol Sci. 2020 Nov 20;21(22):8767. doi: 10.3390/ijms21228767. Int J Mol Sci. 2020. PMID: 33233502 Free PMC article. Review.
-
miRNAs Identify Shared Pathways in Alzheimer's and Parkinson's Diseases.Trends Mol Med. 2019 Aug;25(8):662-672. doi: 10.1016/j.molmed.2019.05.006. Epub 2019 Jun 17. Trends Mol Med. 2019. PMID: 31221572 Review.
-
The relationship between oxidative/nitrative stress and pathological inclusions in Alzheimer's and Parkinson's diseases.Free Radic Biol Med. 2002 Jun 15;32(12):1264-75. doi: 10.1016/s0891-5849(02)00804-3. Free Radic Biol Med. 2002. PMID: 12057764 Review.
-
Shared mechanisms of neurodegeneration in Alzheimer's disease and Parkinson's disease.Biomed Res Int. 2014;2014:648740. doi: 10.1155/2014/648740. Epub 2014 May 12. Biomed Res Int. 2014. PMID: 24900975 Free PMC article. Review.
Cited by
-
Glial Cell Reprogramming in Ischemic Stroke: A Review of Recent Advancements and Translational Challenges.Transl Stroke Res. 2025 Oct;16(5):1811-1835. doi: 10.1007/s12975-025-01331-7. Epub 2025 Feb 4. Transl Stroke Res. 2025. PMID: 39904845 Review.
-
Oxidative damage in neurodegeneration: roles in the pathogenesis and progression of Alzheimer disease.Physiol Rev. 2024 Jan 1;104(1):103-197. doi: 10.1152/physrev.00030.2022. Physiol Rev. 2024. PMID: 37843394 Free PMC article. Review.
-
Significance of Brain Glucose Hypometabolism, Altered Insulin Signal Transduction, and Insulin Resistance in Several Neurological Diseases.Front Endocrinol (Lausanne). 2022 May 9;13:873301. doi: 10.3389/fendo.2022.873301. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35615716 Free PMC article. Review.
-
Investigating the neuroprotective effects of Dracocephalum moldavica extract and its effect on metabolomic profile of rat model of sporadic Alzheimer's disease.Heliyon. 2025 Feb 1;11(3):e42412. doi: 10.1016/j.heliyon.2025.e42412. eCollection 2025 Feb 15. Heliyon. 2025. PMID: 39981356 Free PMC article.
-
Transcriptome analysis reveals organ-specific effects of 2-deoxyglucose treatment in healthy mice.PLoS One. 2024 Mar 7;19(3):e0299595. doi: 10.1371/journal.pone.0299595. eCollection 2024. PLoS One. 2024. PMID: 38451972 Free PMC article.
References
-
- Silverman D.H.S., Chen W., Czernin J., Kowell A.P., Gambhir S.S., Phelps M.E., Lu C.S., Kung de Aburto M.A.K., Chang C.Y., Small G.W., et al. Positron Emission Tomography in Evaluation of Dementia: Regional Brain Metabolism and Long-term Outcome. JAMA. 2001;286:2120–2127. doi: 10.1001/jama.286.17.2120. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
