

Clinical disease characteristics of patients with Niemann-Pick Disease Type C: findings from the International Niemann-Pick Disease Registry (INPDR)
- PMID: 35164809
- PMCID: PMC8842861
- DOI: 10.1186/s13023-022-02200-4
Clinical disease characteristics of patients with Niemann-Pick Disease Type C: findings from the International Niemann-Pick Disease Registry (INPDR)
Abstract
Background: Niemann-Pick Disease Type C (NPC) is an autosomal recessive rare disease characterised by progressive neurovisceral manifestations. The collection of on-going large-scale NPC clinical data may generate better understandings of the natural history of the disease. Here we report NPC patient data from the International Niemann-Pick Disease Registry (INPDR).
Method: The INPDR is a web-based, patient-led independent registry for the collection of prospective and retrospective clinical data from Niemann-Pick Disease patients. Baseline data from NPC patients enrolled into the INPDR from September 2014 to December 2019 was extracted to analyse the demographic, genetic and clinical features of the disease.
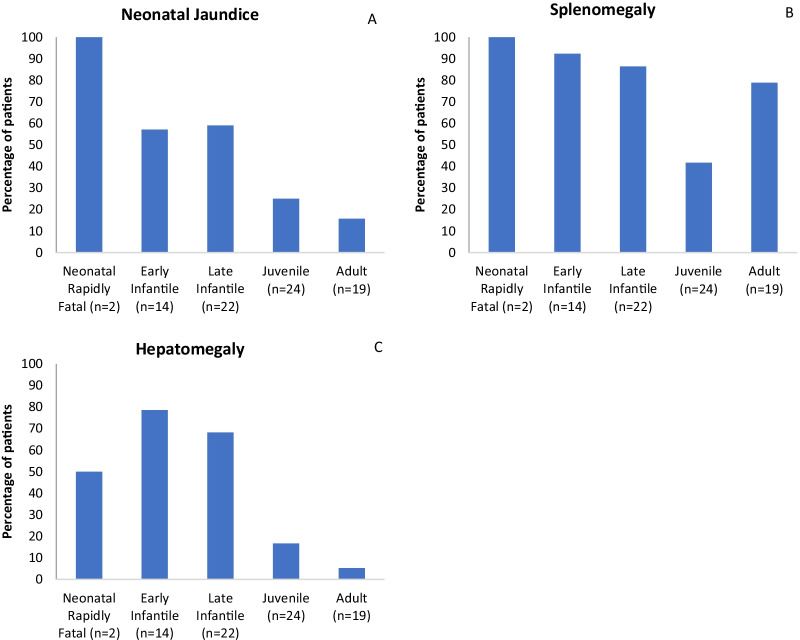
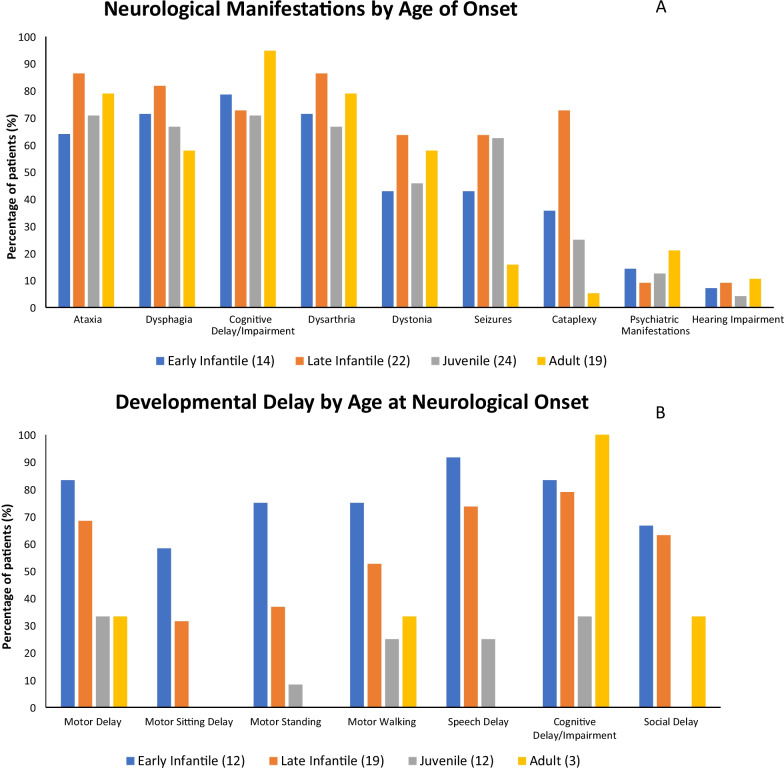
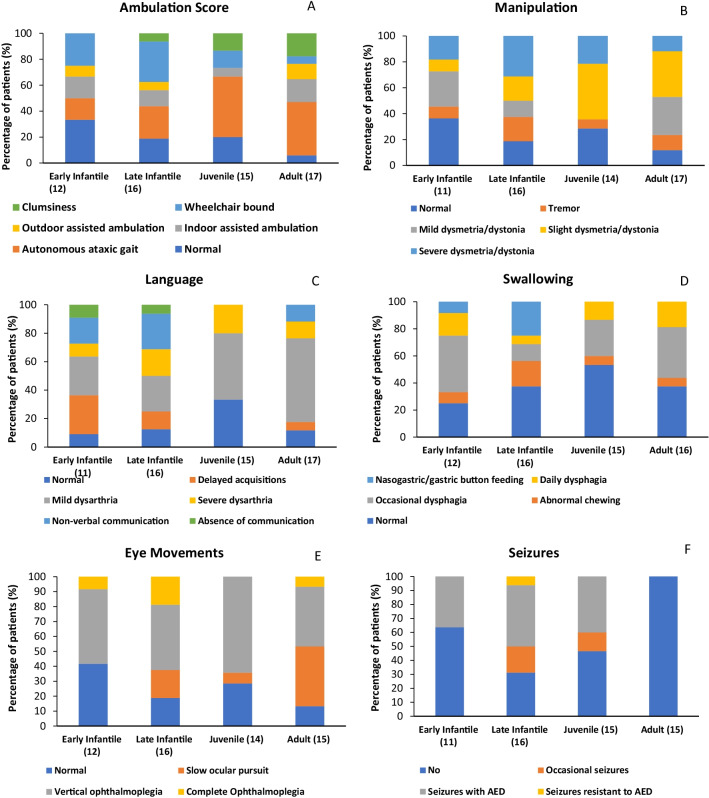
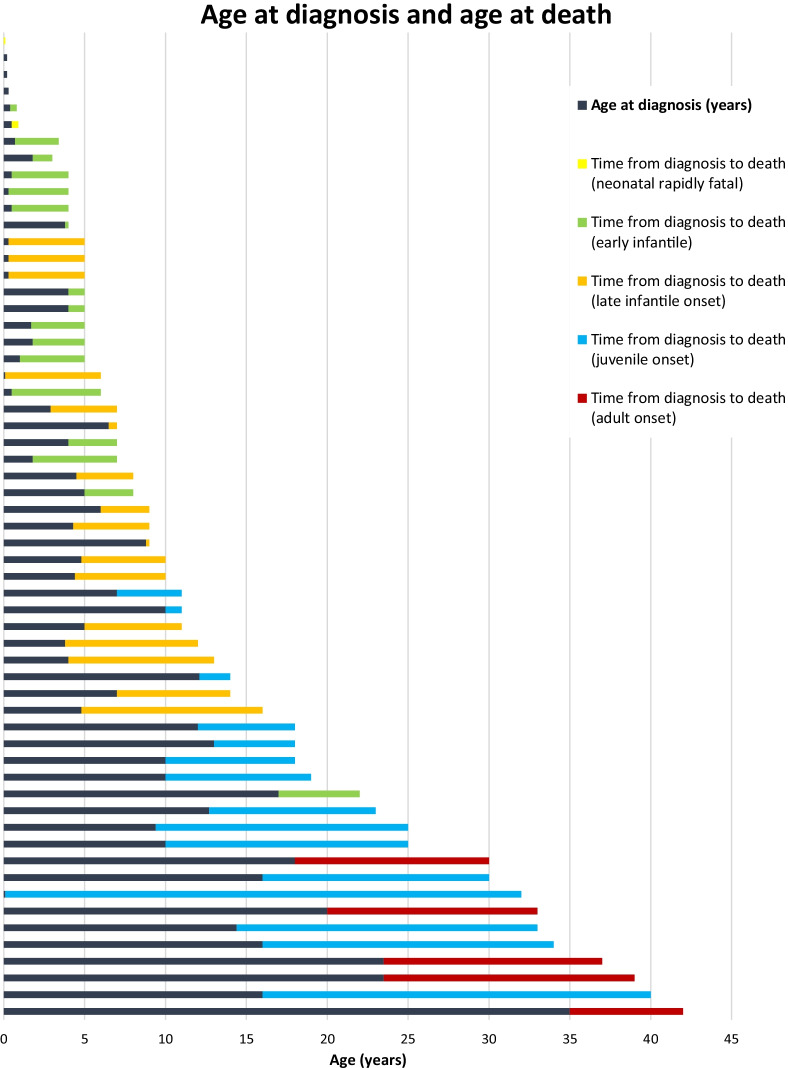
Results: A total of 203 NPC patients from six European countries were included in this study. The mean age (SD) at diagnosis was 11.2 years (14.2). Among enrolled patients, 168 had known neurological manifestations: 43 (24.2%) had early-infantile onset, 47 (26.4%) had late-infantile onset, 41 (23.0%) had juvenile onset, and 37 (20.8%) had adult onset. 10 (5.6%) patients had the neonatal rapidly fatal systemic form. Among the 97 patients with identified NPC1 variants, the most common variant was the c. 3182T > C variant responsible for the p.lle1061Thr protein change, reported in 35.1% (N = 34) of patients. The frequencies of hepatomegaly and neonatal jaundice were greatest in patients with early-infantile and late-infantile neurological onset. Splenomegaly was the most commonly reported observation, including 80% of adult-onset patients. The most commonly reported neurological manifestations were cognitive impairment (78.5%), dysarthria (75.9%), ataxia (75.9%), vertical supranuclear gaze palsy (70.9%) and dysphagia (69.6%). A 6-domain composite disability scale was used to calculate the overall disability score for each neurological form. Across all with neurological onset, the majority of patients showed moderate to severe impairments in all domains, except for 'swallowing' and 'seizure'. The age at diagnosis and death increased with increased age of neurological symptom onset. Miglustat use was recorded in 62.4% of patients and the most common symptomatic therapies used by patients were antiepileptics (32.9%), antidepressants (11.8%) and antacids (9.4%).
Conclusion: The proportion of participants at each age of neurological onset was relatively equal across the cohort. Neurological manifestations, such as ataxia, dysphagia, and dysarthria, were frequently observed across all age categories.
© 2022. The Author(s).
Conflict of interest statement
AD, BB, TH, SJ, PG and MTV has received travel grant from Actelion. MP has received travel grant from Actelion, Biomarin, Orphazyme, Esteve, Janssen and Chiesi. SCB has received travel grant from Amicus Therapeutics and Sanofi Genzyme. Takeda has provided funding to TH, SJ, MTV, AD, BB and MP. Genzyme has provided funding to TH, YN, SJ, SS, JI, MH and MP. Orphazyme has provided funding to JI, AD and BB. Alexion has provided funding to MP. Vtesse has provided travel grants to MTV and MP. MP has received funding from Agio, Amicus and Novartis. SJ has received funding from Biomarin, Ultragenyx, PTC and Orchard Theraputics.
Figures
References
-
- Hammond N, Munkacsi A, Sturley S. The complexity of a monogenic neurodegenerative disease: More than two decades of therapeutic driven research into Niemann-Pick type C disease. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids. 2019;164(8):1109–1123. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
