

Ethylmalonic encephalopathy masquerading as meningococcemia
- PMID: 35165146
- PMCID: PMC8958906
- DOI: 10.1101/mcs.a006193
Ethylmalonic encephalopathy masquerading as meningococcemia
Abstract
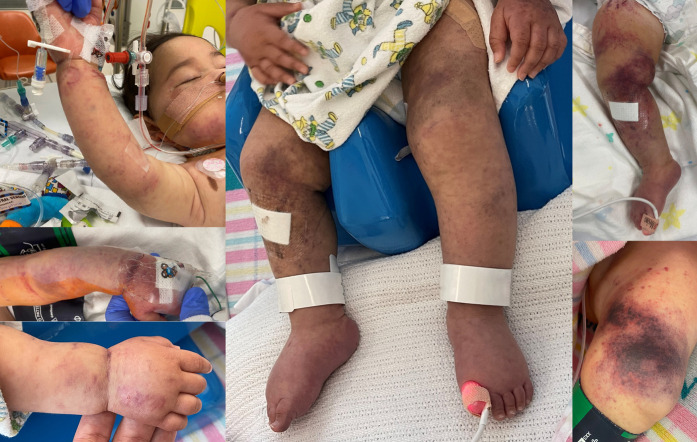
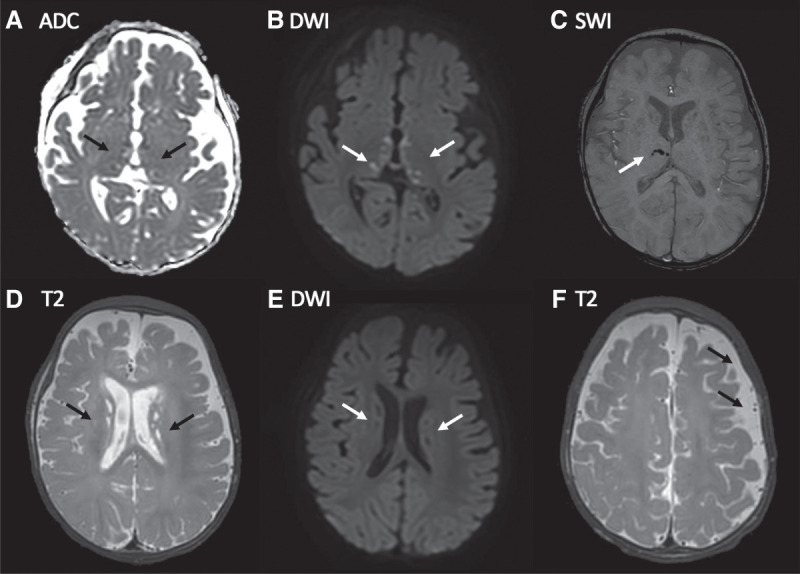
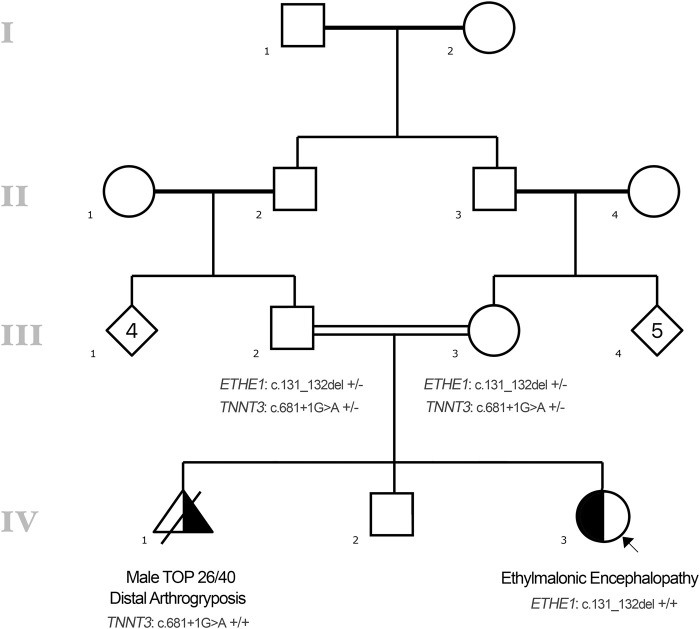
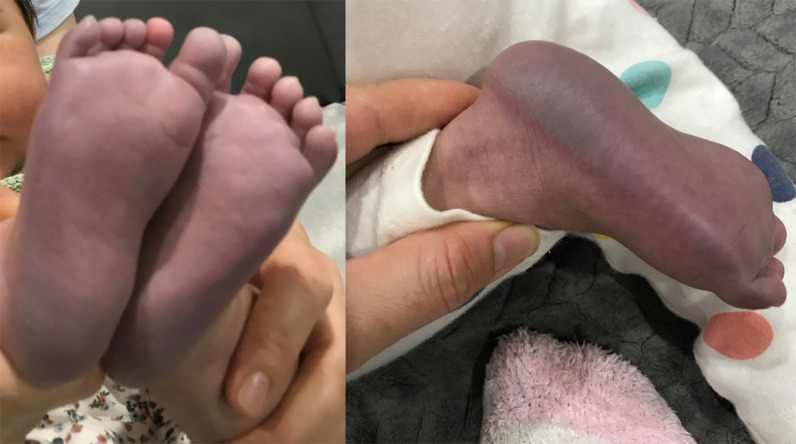
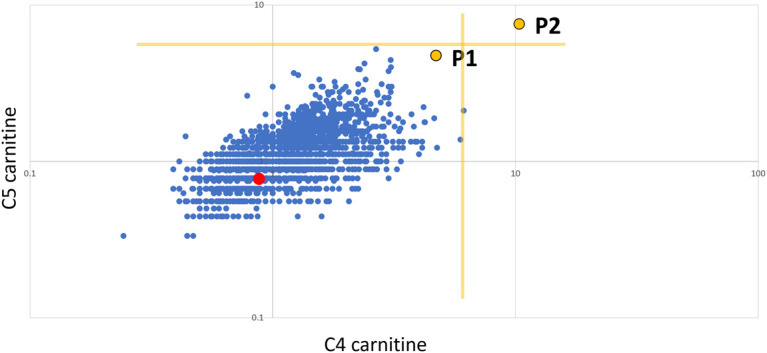
Ethylmalonic encephalopathy (MIM #602473) is a rare autosomal recessive metabolic condition caused by biallelic variants in ETHE1 (MIM #608451), characterized by global developmental delay, infantile hypotonia, seizures, and microvascular damage. The microvascular changes result in a pattern of relapsing spontaneous diffuse petechiae and purpura, positional acrocyanosis, and pedal edema, hemorrhagic suffusions of mucous membranes, and chronic diarrhea. Here, we describe an instructive case in which ethylmalonic encephalopathy masqueraded as meningococcal septicemia and shock. Ultrarapid whole-genome testing (time to result 60 h) and prompt biochemical analysis facilitated accurate diagnosis and counseling with rapid implementation of precision treatment for the metabolic crisis related to this condition. This case provides a timely reminder to consider rare genetic diagnoses when atypical features of more common conditions are present, with an early referral to ensure prompt biochemical and genomic diagnosis.
Keywords: acrocyanosis; ankle clonus; central hypotonia; cerebral ischemia; delayed fine motor development; delayed gross motor development; ethylmalonic aciduria; petechiae; progressive encephalopathy; recurrent cerebral hemorrhage.
© 2022 Horton et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Blau N, Duran M, Gibson KM, Dionisi-Vici C. 2014. Physician's guide to the diagnosis, treatment, and follow-up of inherited metabolic diseases. Springer-Verlag, Berlin.
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical