

Phenotypic and Genomic Diversification in Complex Carbohydrate-Degrading Human Gut Bacteria
- PMID: 35166563
- PMCID: PMC8845570
- DOI: 10.1128/msystems.00947-21
Phenotypic and Genomic Diversification in Complex Carbohydrate-Degrading Human Gut Bacteria
Abstract
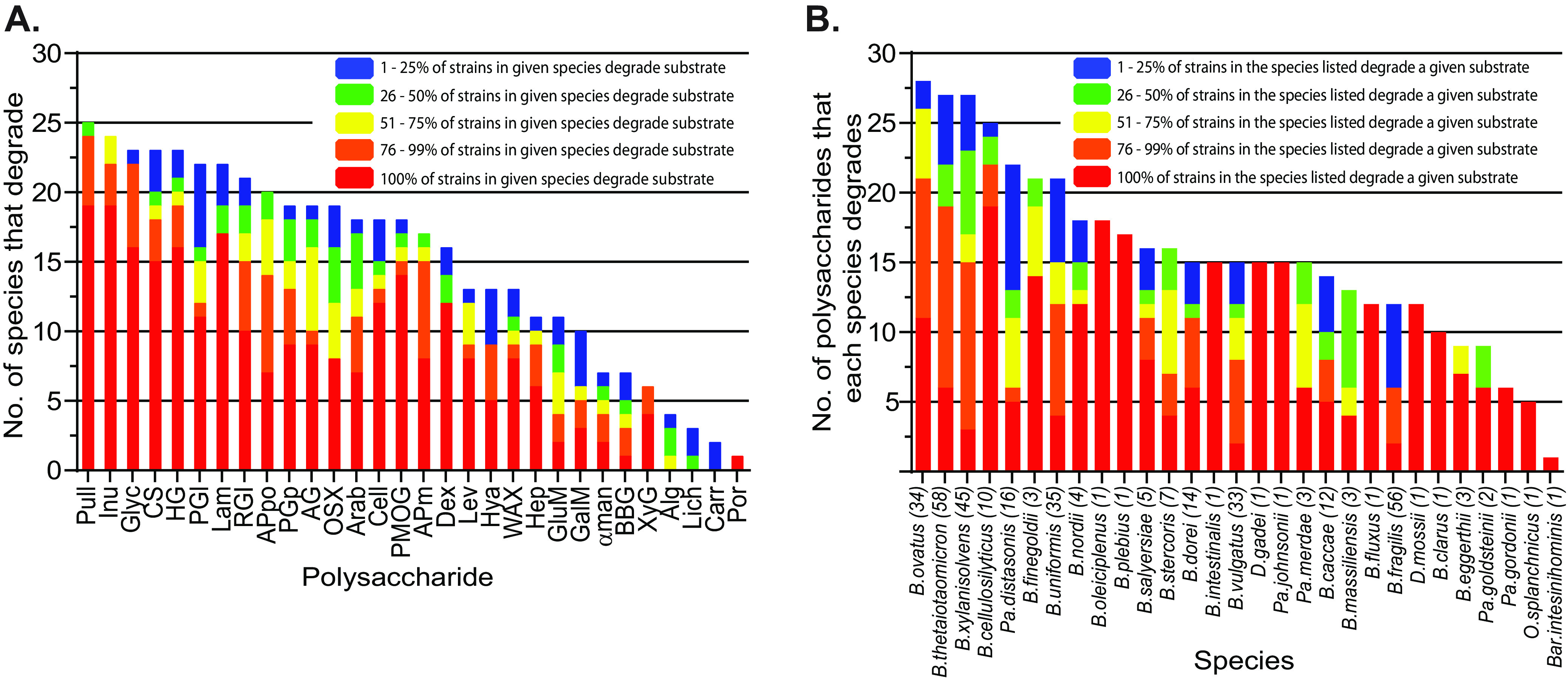
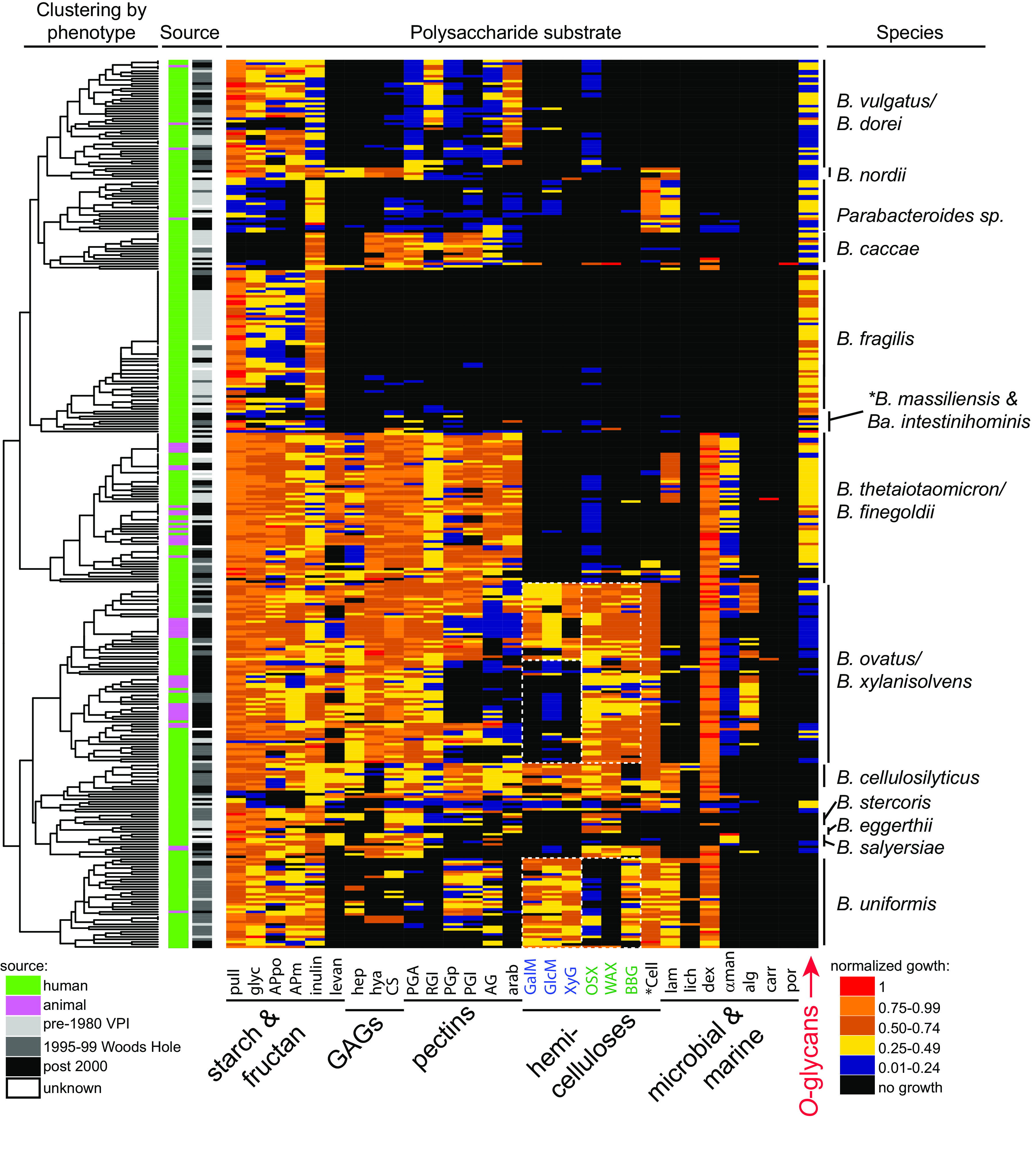
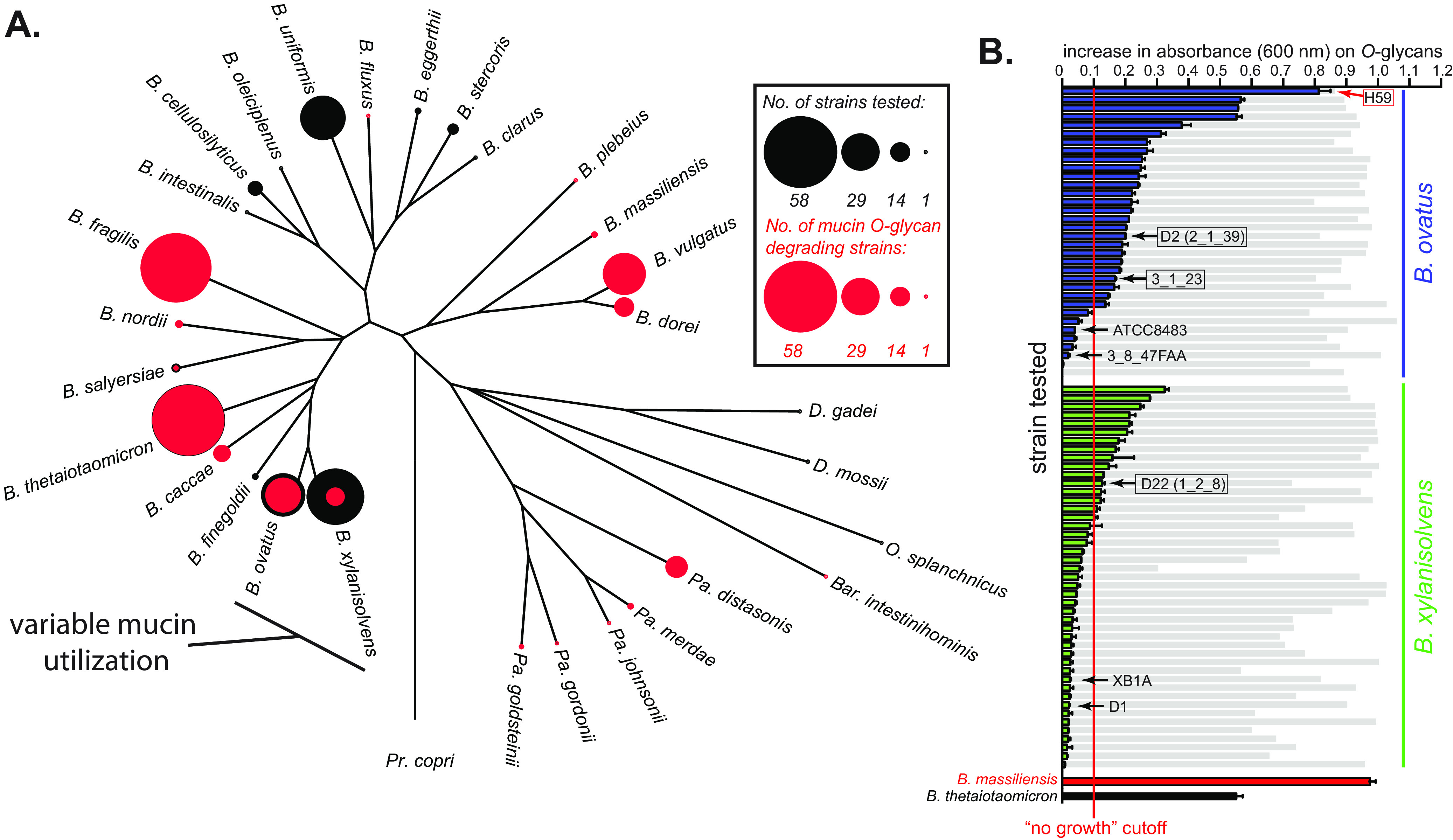
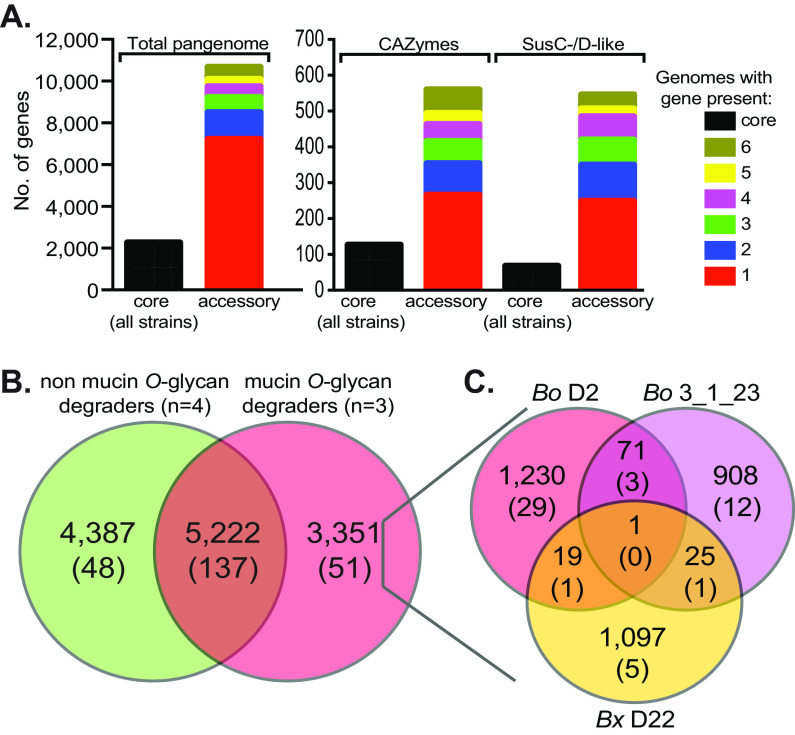
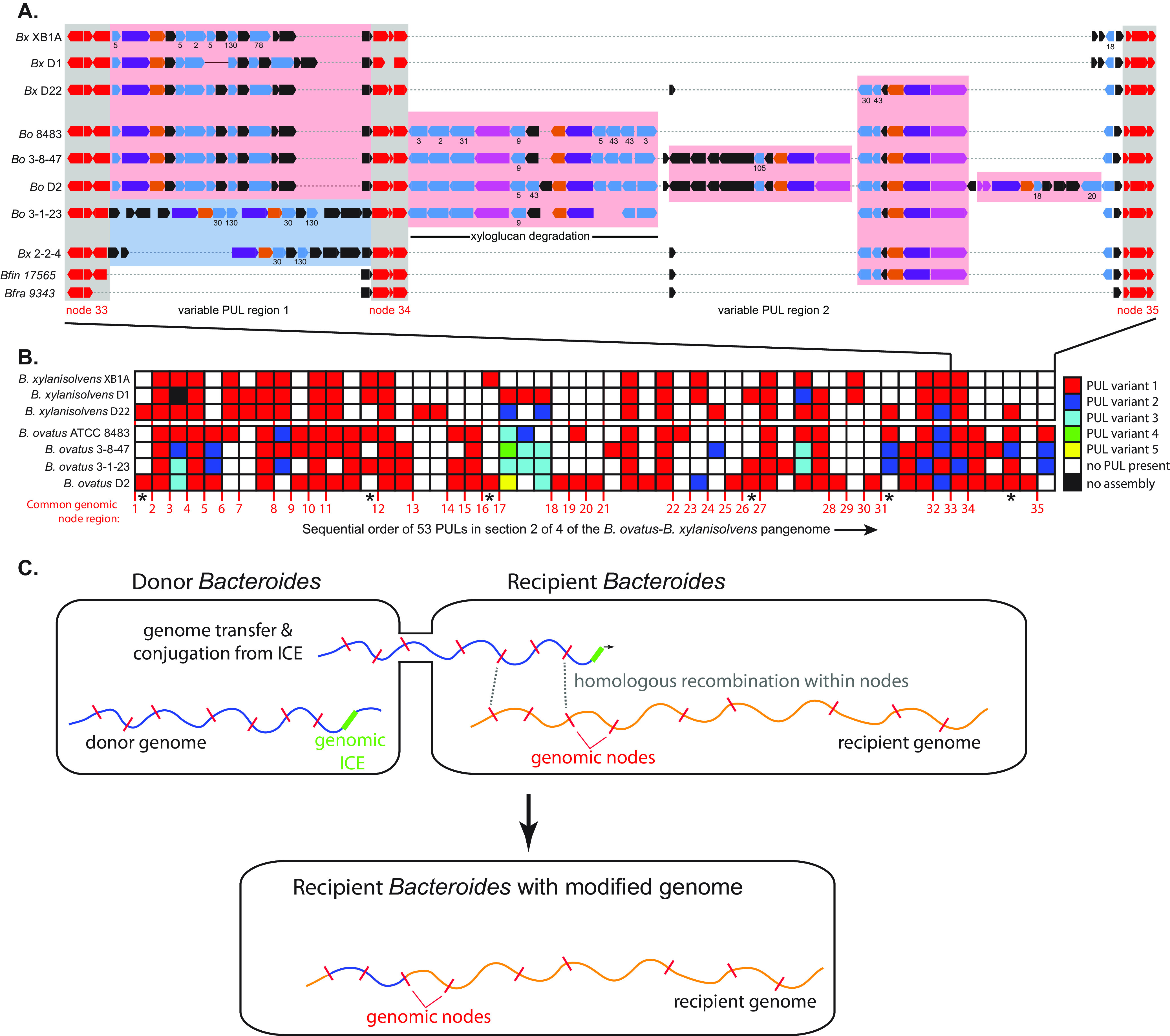
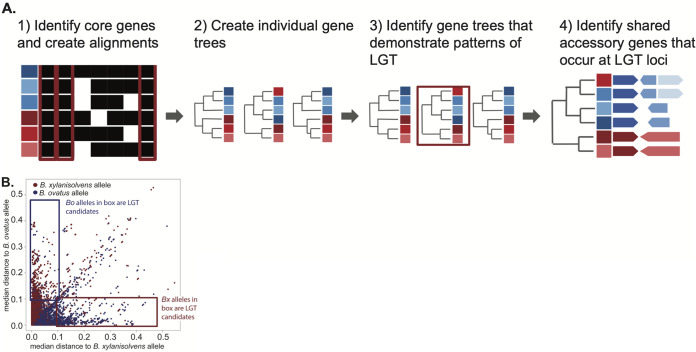
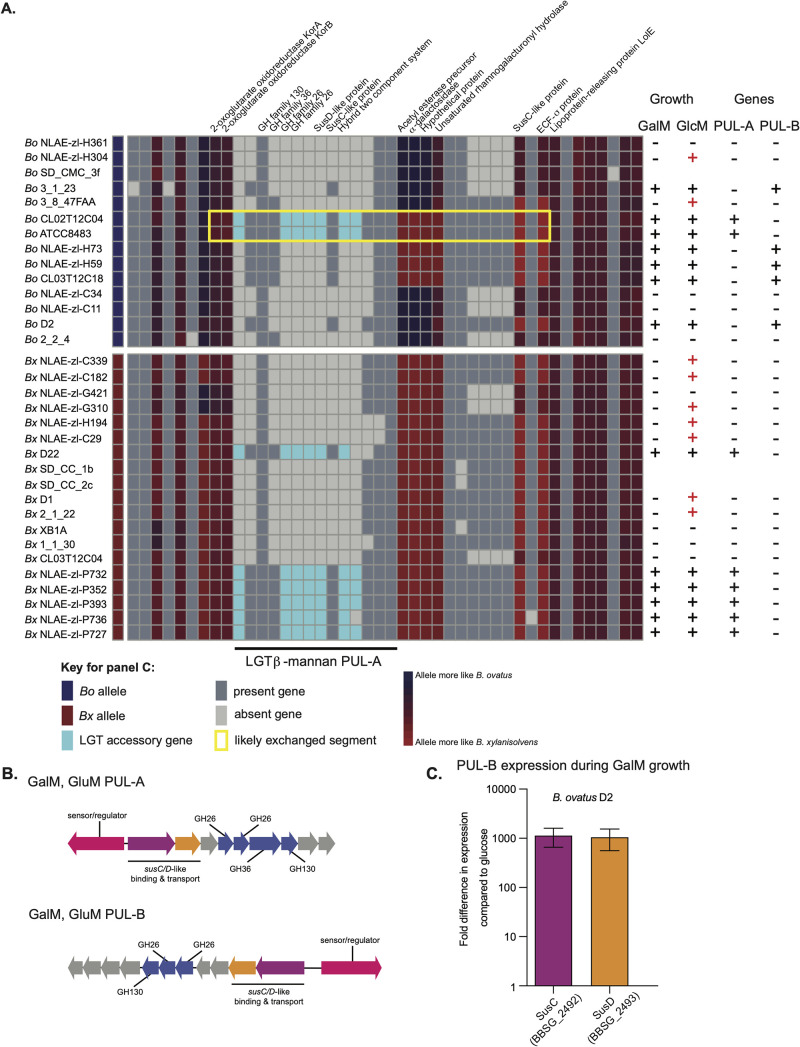
Symbiotic bacteria are responsible for the majority of complex carbohydrate digestion in the human colon. Since the identities and amounts of dietary polysaccharides directly impact the gut microbiota, determining which microorganisms consume specific nutrients is central for defining the relationship between diet and gut microbial ecology. Using a custom phenotyping array, we determined carbohydrate utilization profiles for 354 members of the Bacteroidetes, a dominant saccharolytic phylum. There was wide variation in the numbers and types of substrates degraded by individual bacteria, but phenotype-based clustering grouped members of the same species indicating that each species performs characteristic roles. The ability to utilize dietary polysaccharides and endogenous mucin glycans was negatively correlated, suggesting exclusion between these niches. By analyzing related Bacteroides ovatus/Bacteroides xylanisolvens strains that vary in their ability to utilize mucin glycans, we addressed whether gene clusters that confer this complex, multilocus trait are being gained or lost in individual strains. Pangenome reconstruction of these strains revealed a remarkably mosaic architecture in which genes involved in polysaccharide metabolism are highly variable and bioinformatics data provide evidence of interspecies gene transfer that might explain this genomic heterogeneity. Global transcriptomic analyses suggest that the ability to utilize mucin has been lost in some lineages of B. ovatus and B. xylanisolvens, which harbor residual gene clusters that are involved in mucin utilization by strains that still actively express this phenotype. Our data provide insight into the breadth and complexity of carbohydrate metabolism in the microbiome and the underlying genomic events that shape these behaviors. IMPORTANCE Nonharmful bacteria are the primary microbial symbionts that inhabit the human gastrointestinal tract. These bacteria play many beneficial roles and in some cases can modify disease states, making it important to understand which nutrients sustain specific lineages. This knowledge will in turn lead to strategies to intentionally manipulate the gut microbial ecosystem. We designed a scalable, high-throughput platform for measuring the ability of gut bacteria to utilize polysaccharides, of which many are derived from dietary fiber sources that can be manipulated easily. Our results provide paths to expand phenotypic surveys of more diverse gut bacteria to understand their functions and also to leverage dietary fibers to alter the physiology of the gut microbial community.
Keywords: Bacteroides; microbiome; pangenome; polysaccharides.
Conflict of interest statement
The authors declare no conflict of interest.
We declare no competing interests.
Figures
Comment in
-
Mind the Gap: Bridging the Divide from Sequencing Data to Empiric Phenotypes in the Human Gut Microbiota.mSystems. 2022 Jun 28;7(3):e0020722. doi: 10.1128/msystems.00207-22. Epub 2022 Jun 13. mSystems. 2022. PMID: 35695430 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
