

DNA methylation cues in nucleosome geometry, stability and unwrapping
- PMID: 35166834
- PMCID: PMC8881801
- DOI: 10.1093/nar/gkac097
DNA methylation cues in nucleosome geometry, stability and unwrapping
Abstract
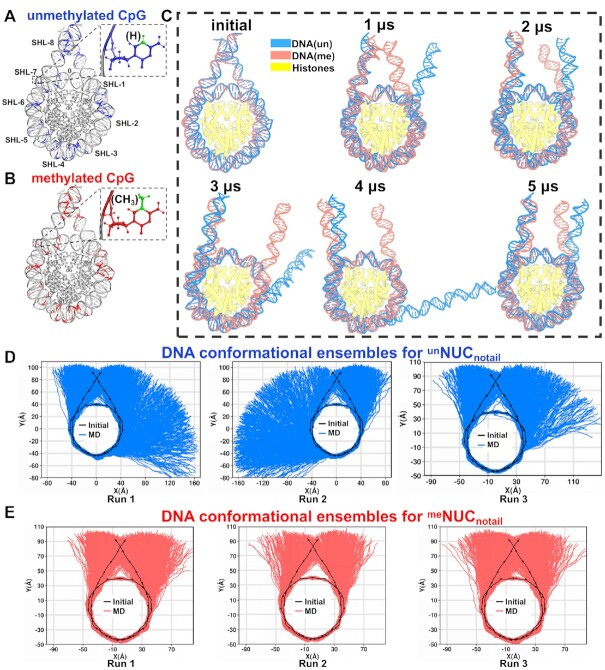
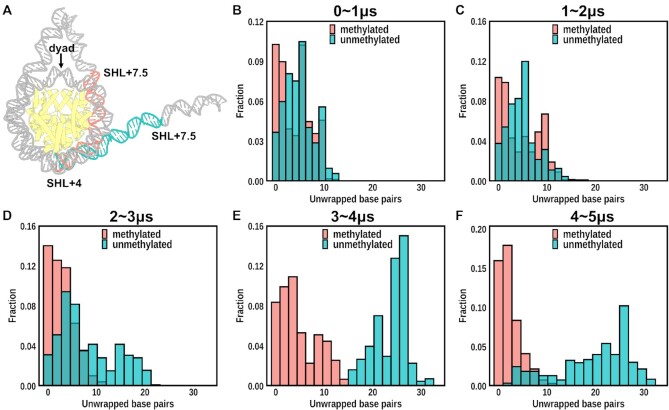
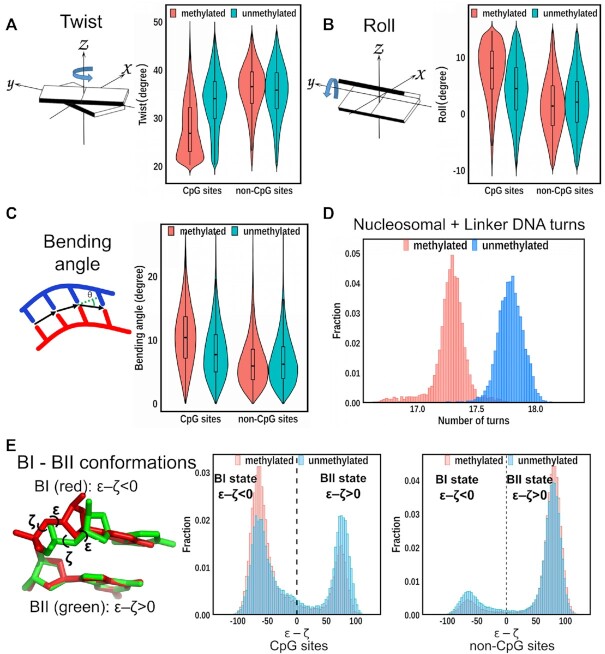
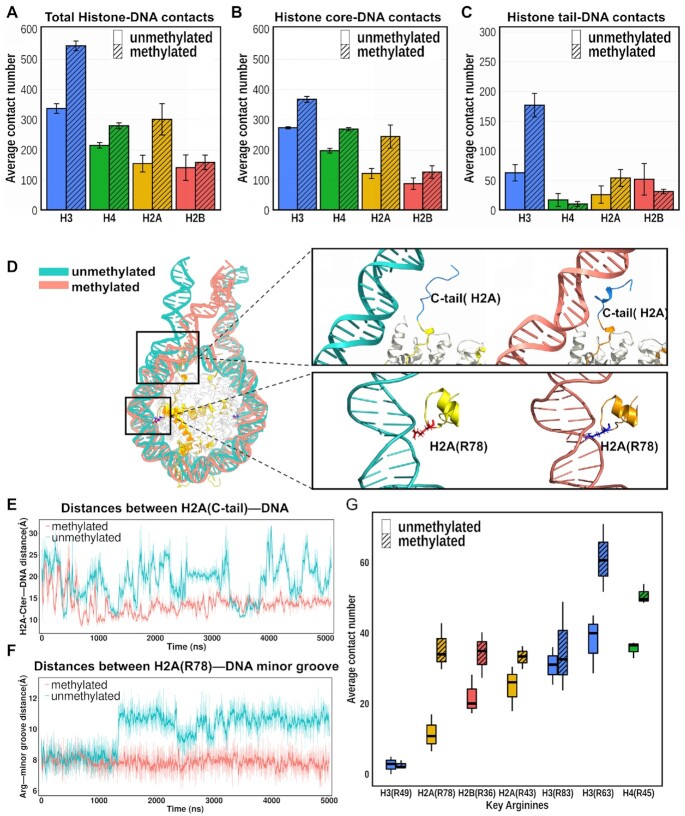
Cytosine methylation at the 5-carbon position is an essential DNA epigenetic mark in many eukaryotic organisms. Although countless structural and functional studies of cytosine methylation have been reported, our understanding of how it influences the nucleosome assembly, structure, and dynamics remains obscure. Here, we investigate the effects of cytosine methylation at CpG sites on nucleosome dynamics and stability. By applying long molecular dynamics simulations on several microsecond time scale, we generate extensive atomistic conformational ensembles of full nucleosomes. Our results reveal that methylation induces pronounced changes in geometry for both linker and nucleosomal DNA, leading to a more curved, under-twisted DNA, narrowing the adjacent minor grooves, and shifting the population equilibrium of sugar-phosphate backbone geometry. These DNA conformational changes are associated with a considerable enhancement of interactions between methylated DNA and the histone octamer, doubling the number of contacts at some key arginines. H2A and H3 tails play important roles in these interactions, especially for DNA methylated nucleosomes. This, in turn, prevents a spontaneous DNA unwrapping of 3-4 helical turns for the methylated nucleosome with truncated histone tails, otherwise observed in the unmethylated system on several microseconds time scale.
Published by Oxford University Press on behalf of Nucleic Acids Research 2022.
Figures
References
-
- Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012; 13:484–492. - PubMed
-
- Bergman Y., Cedar H.. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013; 20:274–281. - PubMed
-
- Cedar H., Bergman Y.. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009; 10:295–304. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
