

Whole-genome analyses reveal a novel prophage and cgSNPs-derived sublineages of Brachyspira hyodysenteriae ST196
- PMID: 35168548
- PMCID: PMC8845278
- DOI: 10.1186/s12864-022-08347-5
Whole-genome analyses reveal a novel prophage and cgSNPs-derived sublineages of Brachyspira hyodysenteriae ST196
Abstract
Background: Brachyspira (B.) hyodysenteriae is a fastidious anaerobe spirochete that can cause swine dysentery, a severe mucohaemorragic colitis that affects pig production and animal welfare worldwide. In Switzerland, the population of B. hyodysenteriae is characterized by the predominance of macrolide-lincosamide-resistant B. hyodysenteriae isolates of sequence type (ST) ST196, prompting us to obtain deeper insights into the genomic structure and variability of ST196 using pangenome and whole genome variant analyses.
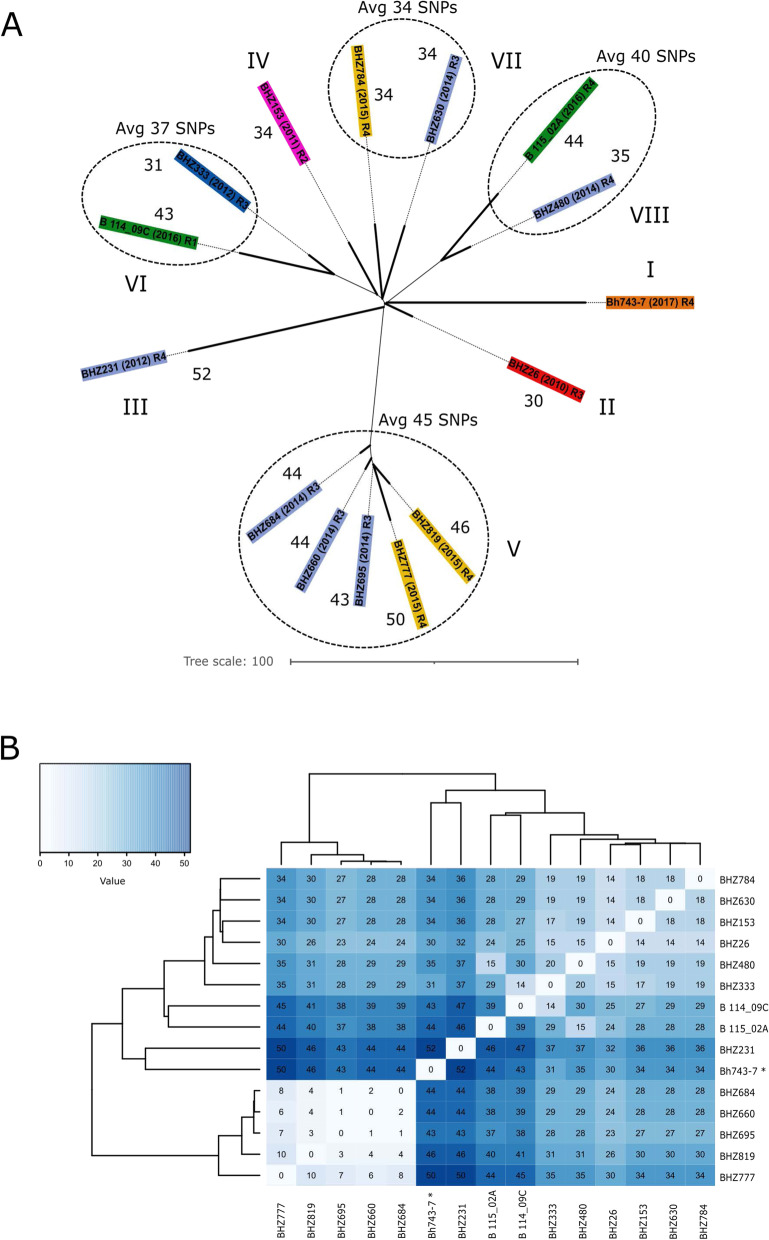
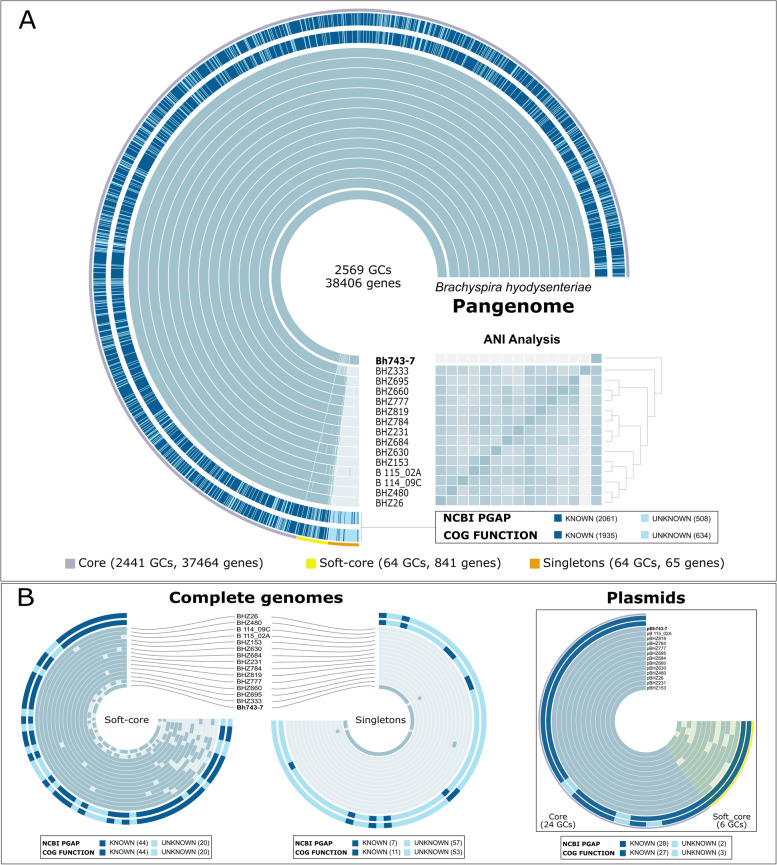
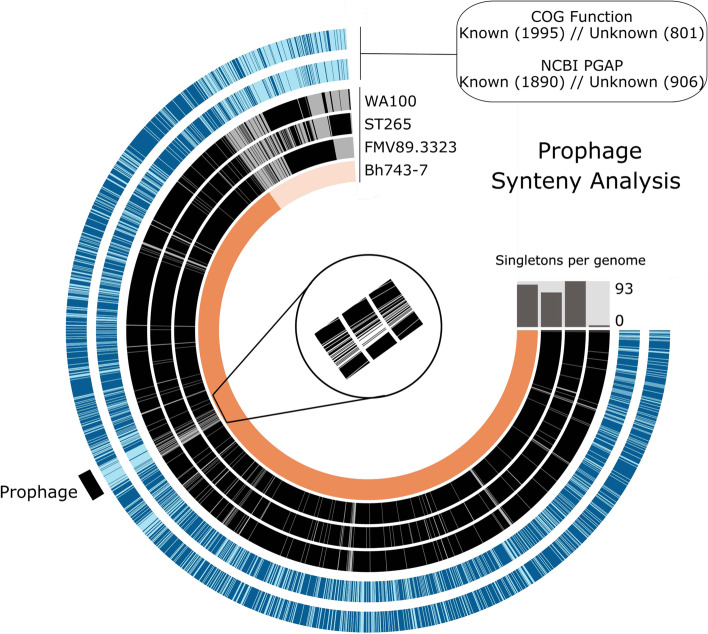
Results: The draft genome of 14 B. hyodysenteriae isolates of ST196, sampled during a 7-year period from geographically distant pig herds, was obtained by whole-genome sequencing (WGS) and compared to the complete genome of the B. hyodysenteriae isolate Bh743-7 of ST196 used as reference. Variability results revealed the existence of 30 to 52 single nucleotide polymorphisms (SNPs), resulting in eight sublineages of ST196. The pangenome analysis led to the identification of a novel prophage, pphBhCH20, of the Siphoviridae family in a single isolate of ST196, which suggests that horizontal gene transfer events may drive changes in genomic structure.
Conclusions: This study contributes to the catalogue of publicly available genomes and provides relevant bioinformatic tools and information for further comparative genomic analyses for B. hyodysenteriae. It reveals that Swiss B. hyodysenteriae isolates of the same ST may have evolved independently over time by point mutations and acquisition of larger genetic elements. In line with this, the third type of mobile genetic element described so far in B. hyodysenteriae, the novel prophage pphBhCH20, has been identified in a single isolate of B. hyodysenteriae of ST196.
Keywords: Bioinformatics; Horizontal-gene transfer; Pangenome; Singletons; Structural variations; Swine dysentery; WGS.
© 2022. The Author(s).
Conflict of interest statement
None of the authors of this paper have a financial or personal relationship with other people or organizations that could inappropriately influence or bias the content of the paper.
Figures

References
-
- Hampson D, Lugsomya K, La T, Dale Phillips N, J. Trott D, Abraham S. Antimicrobial resistance in Brachyspira – An increasing problem for disease control. Vet Microbiol. 2019;229:59-71. - PubMed
-
- De Luca S, Nicholson P, Magistrali CF, García-Martín AB, Rychener L, Zeeh F, Frey J, Perreten V. Corrigendum to “Transposon-associated lincosamide resistance lnu(C) gene identified in Brachyspira hyodysenteriae ST83” [Vet Microbiol. 214 (2018) 51–55]. Vet Microbiol. 2018;220:113. - PubMed
-
- De Luca S, Nicholson P, Magistrali CF, García-Martín AB, Rychener L, Zeeh F, Frey J, Perreten V. Transposon-associated lincosamide resistance lnu(C) gene identified in Brachyspira hyodysenteriae ST83. Vet Microbiol. 2018;214:51–5. - PubMed
