

Angioplasty induces epigenomic remodeling in injured arteries
- PMID: 35169042
- PMCID: PMC8860099
- DOI: 10.26508/lsa.202101114
Angioplasty induces epigenomic remodeling in injured arteries
Abstract
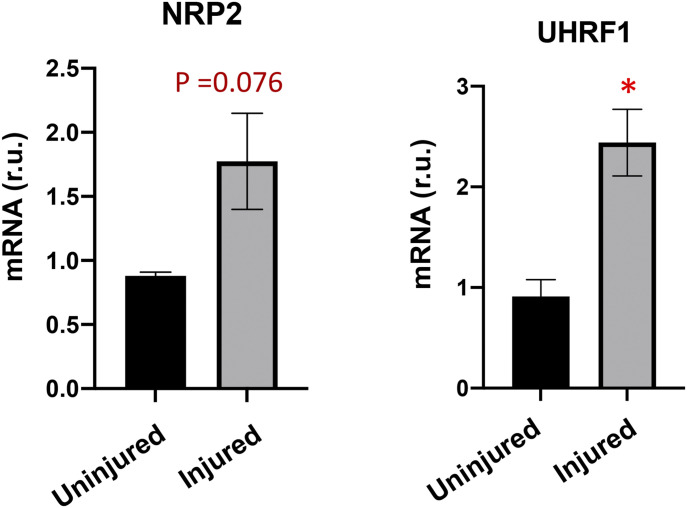
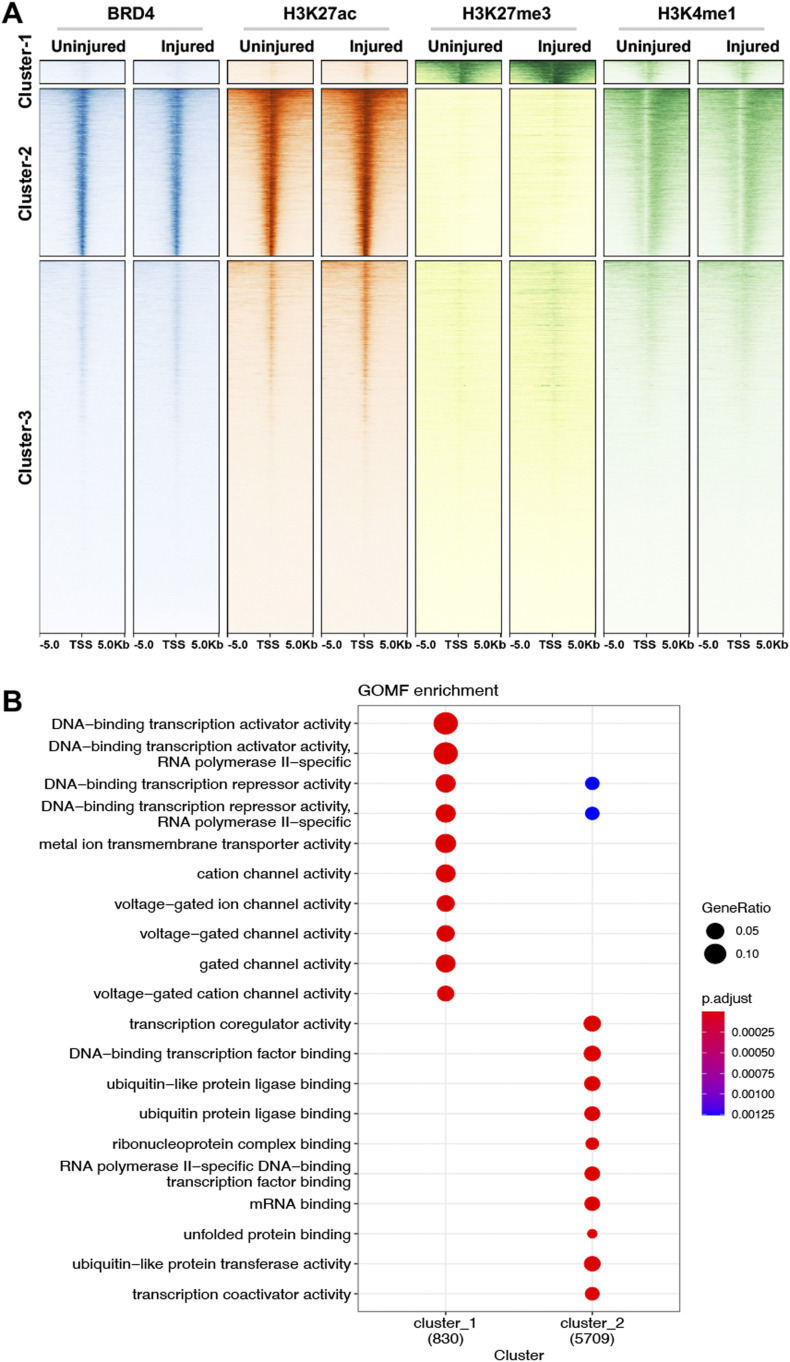
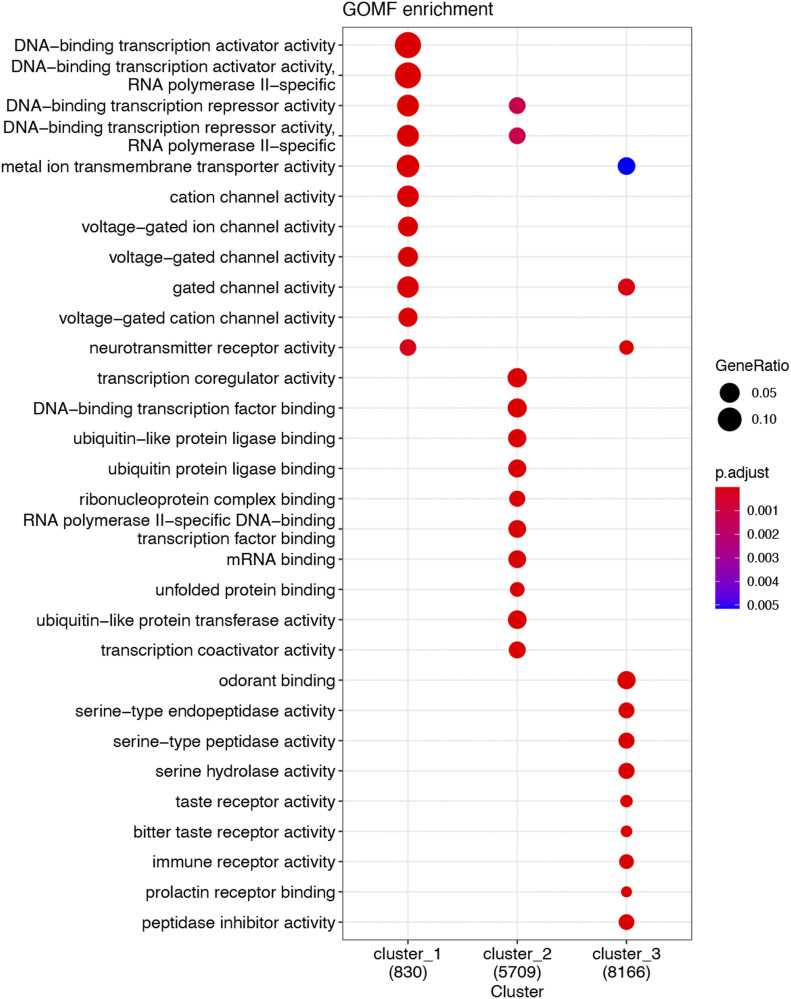
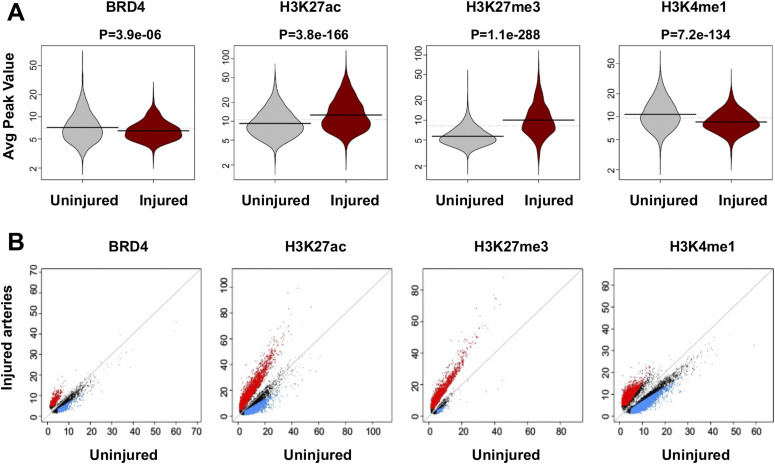
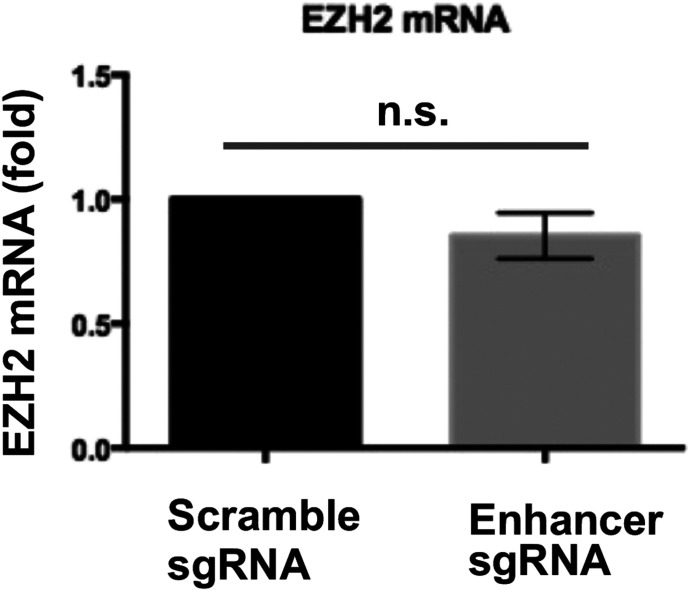
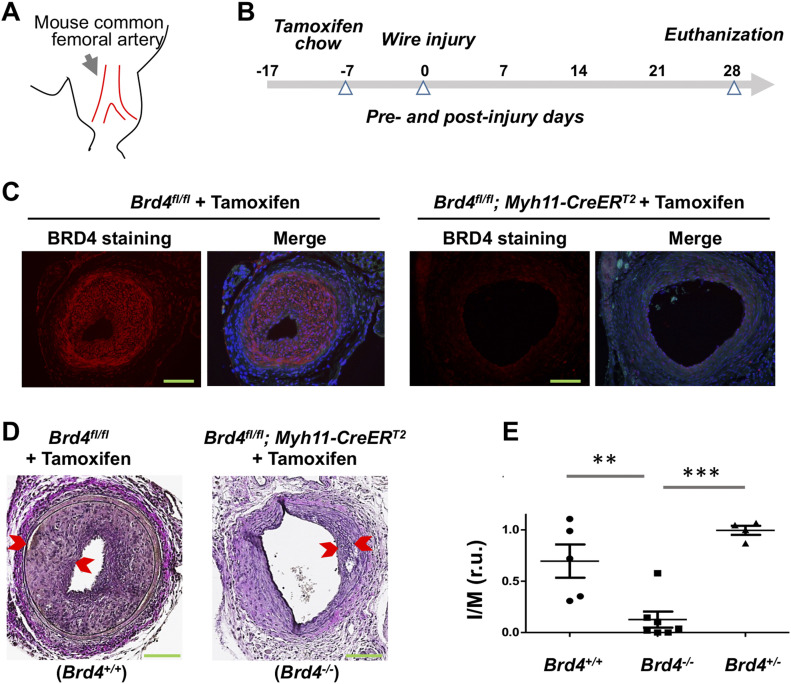
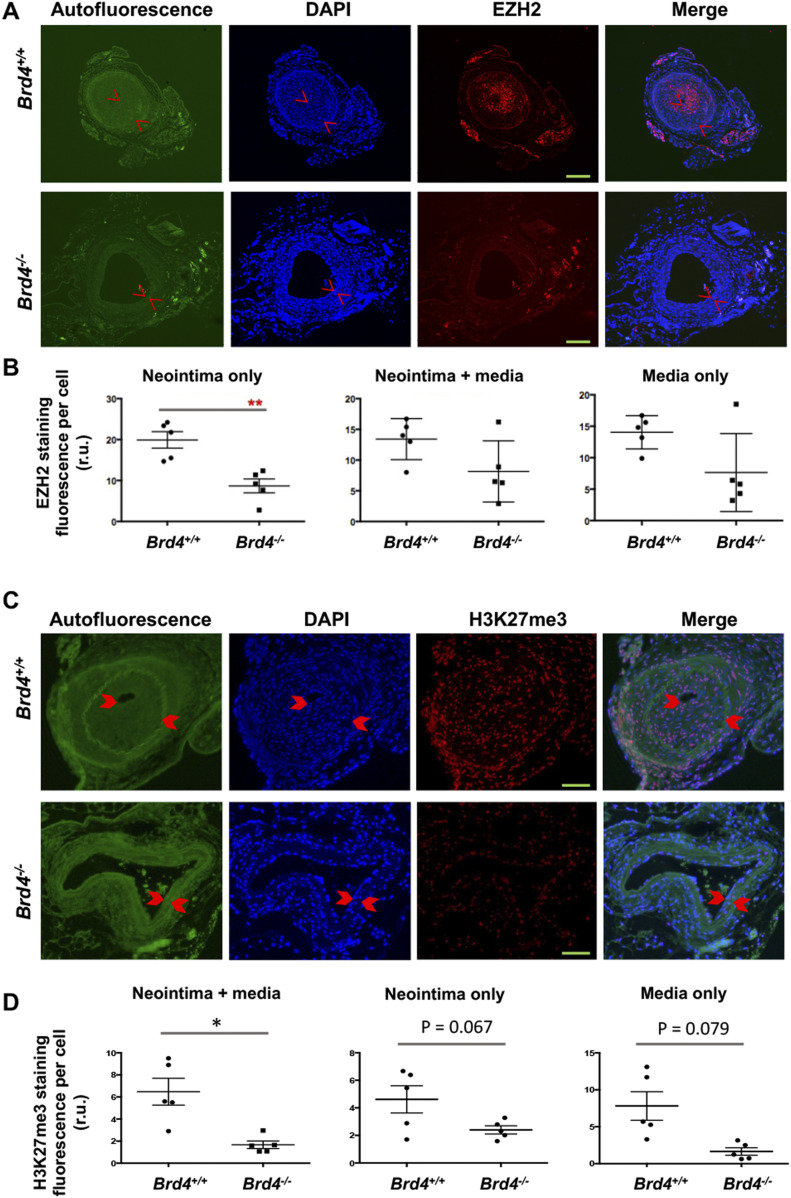
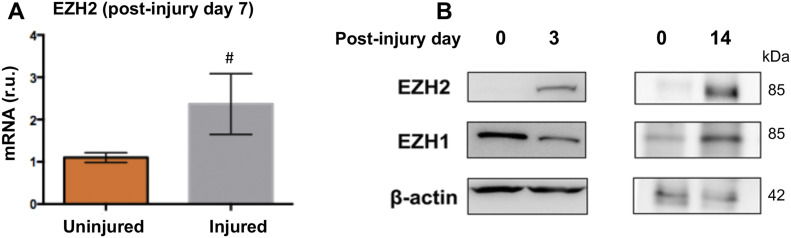
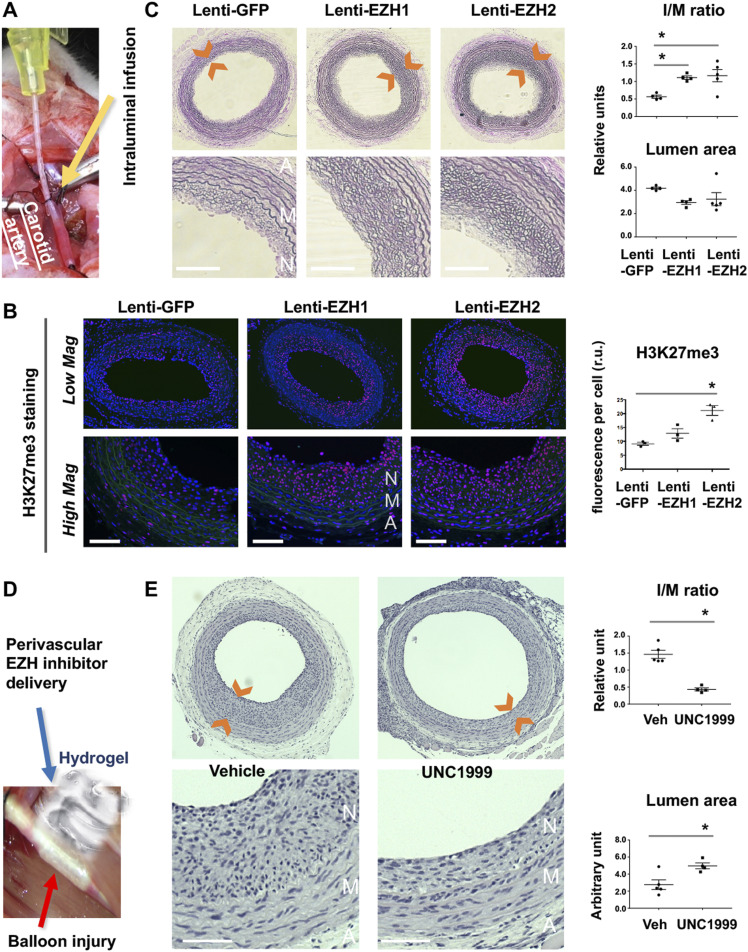
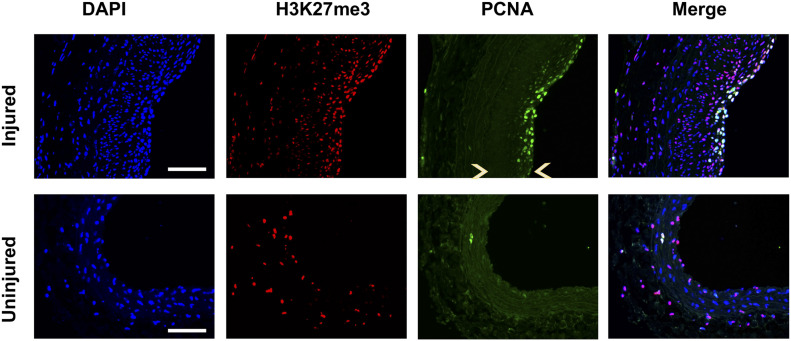
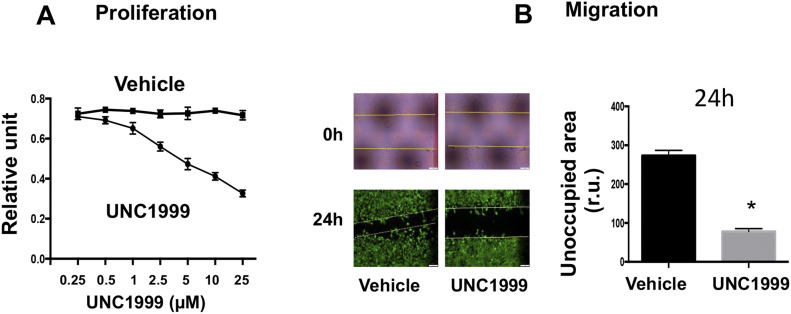
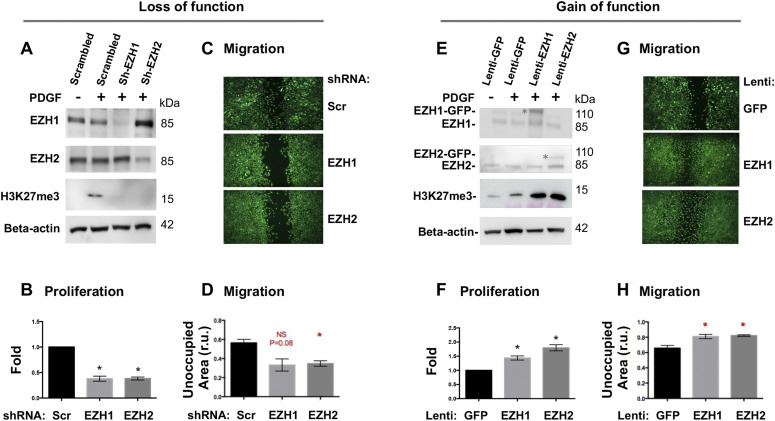
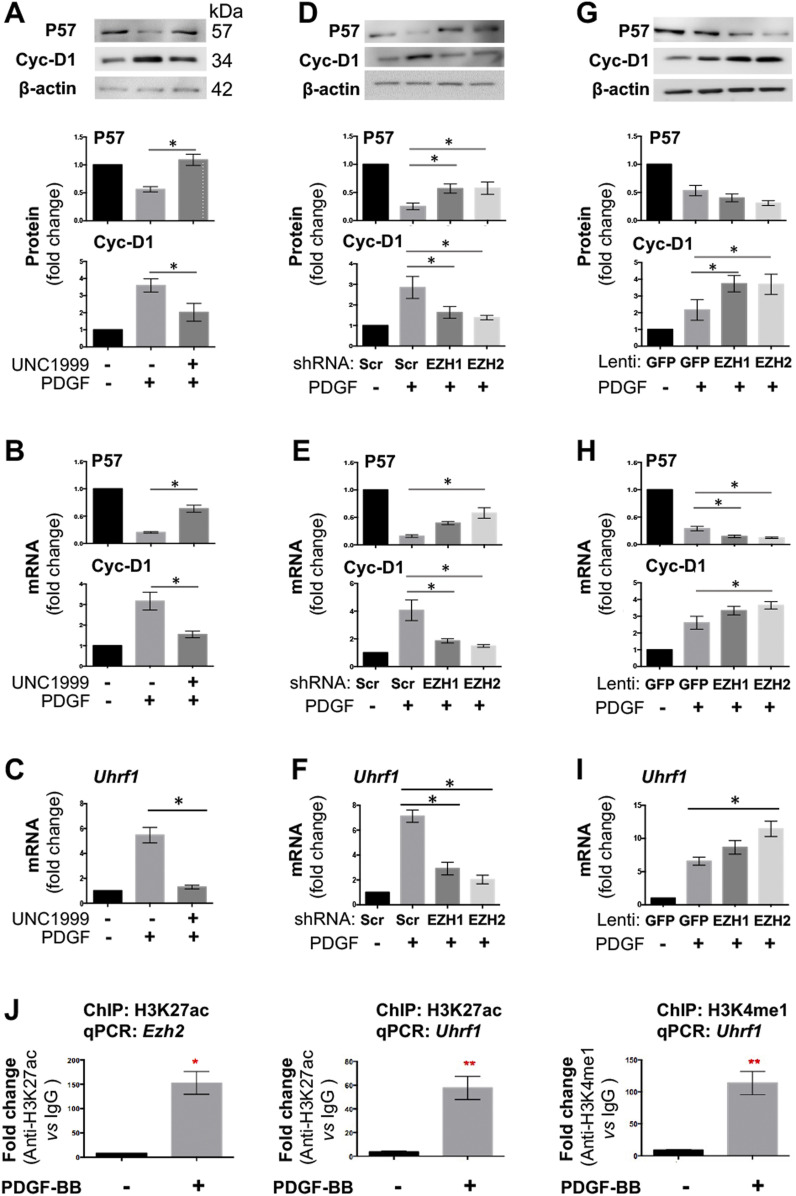
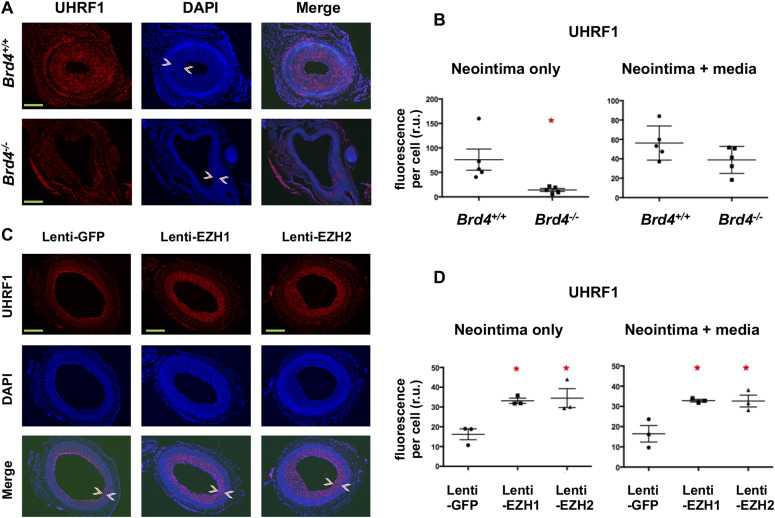
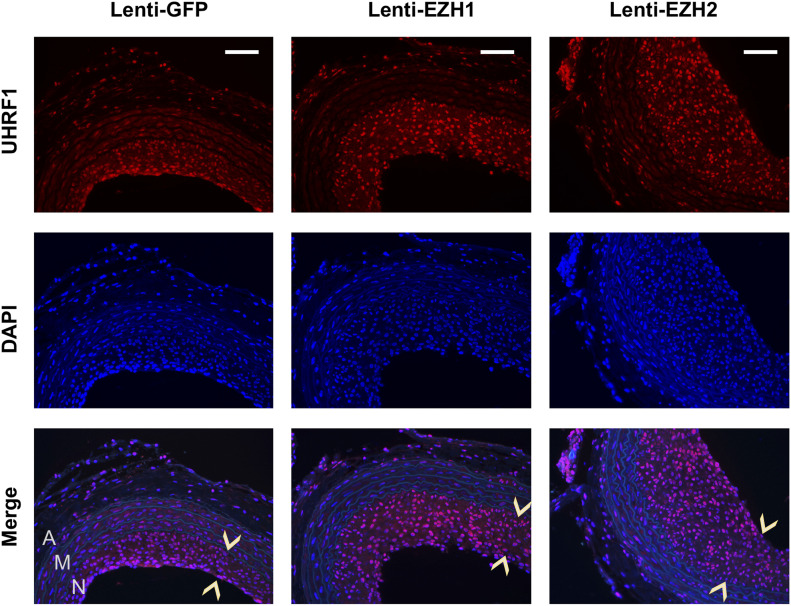
Neointimal hyperplasia/proliferation (IH) is the primary etiology of vascular stenosis. Epigenomic studies concerning IH have been largely confined to in vitro models, and IH-underlying epigenetic mechanisms remain poorly understood. This study integrates information from in vivo epigenomic mapping, conditional knockout, gene transfer and pharmacology in rodent models of IH. The data from injured (IH-prone) rat arteries revealed a surge of genome-wide occupancy by histone-3 lysine-27 trimethylation (H3K27me3), a gene-repression mark. This was unexpected in the traditional view of prevailing post-injury gene activation rather than repression. Further analysis illustrated a shift of H3K27me3 enrichment to anti-proliferative genes, from pro-proliferative genes where gene-activation mark H3K27ac(acetylation) accumulated instead. H3K27ac and its reader BRD4 (bromodomain protein) co-enriched at Ezh2; conditional BRD4 knockout in injured mouse arteries reduced H3K27me3 and its writer EZH2, which positively regulated another pro-IH chromatin modulator UHRF1. Thus, results uncover injury-induced loci-specific H3K27me3 redistribution in the epigenomic landscape entailing BRD4→EZH2→UHRF1 hierarchical regulations. Given that these players are pharmaceutical targets, further research may help improve treatments of IH.
© 2022 Zhang et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
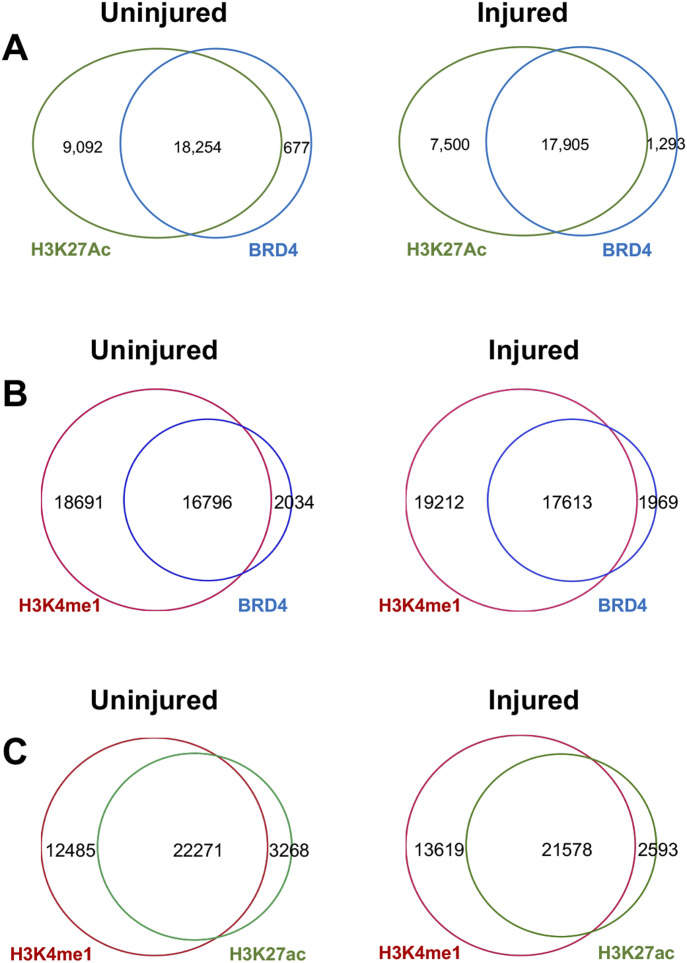
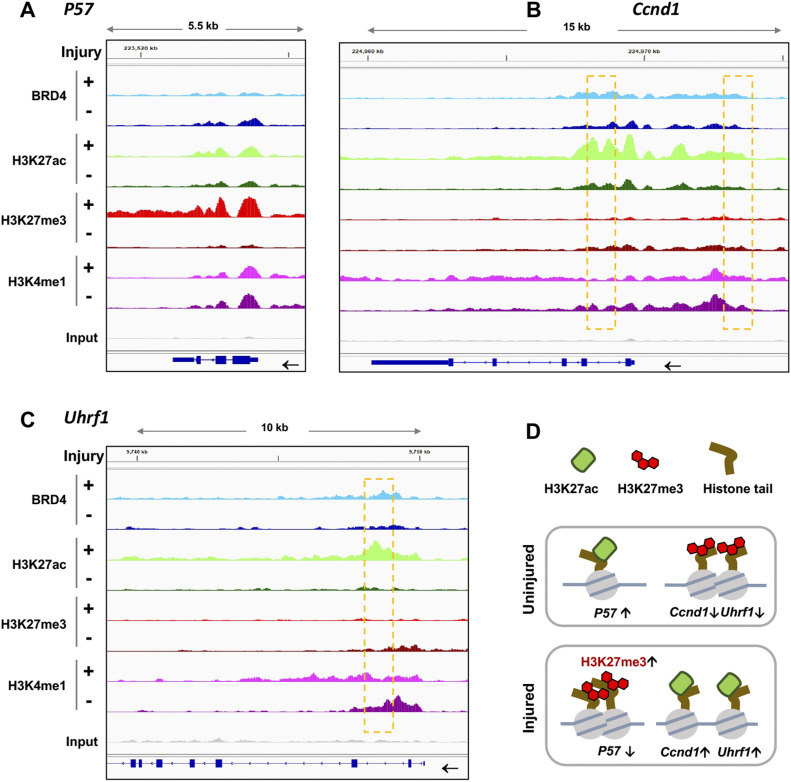
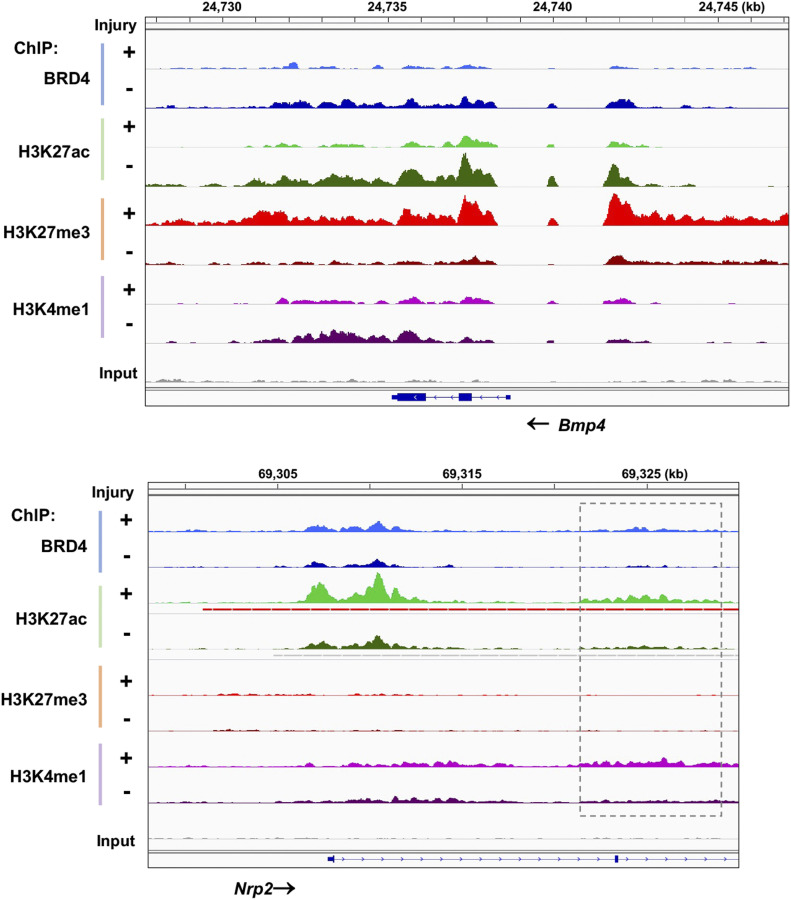
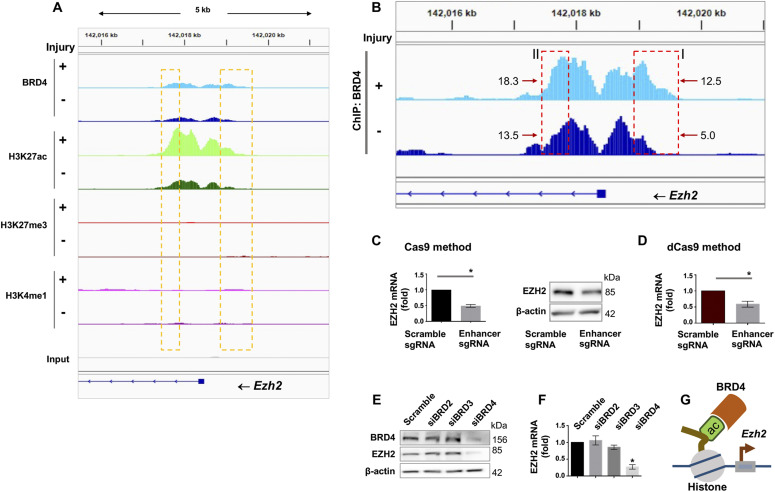
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases