

Human ARHGEF9 intellectual disability syndrome is phenocopied by a mutation that disrupts collybistin binding to the GABAA receptor α2 subunit
- PMID: 35169261
- PMCID: PMC9095487
- DOI: 10.1038/s41380-022-01468-z
Human ARHGEF9 intellectual disability syndrome is phenocopied by a mutation that disrupts collybistin binding to the GABAA receptor α2 subunit
Abstract
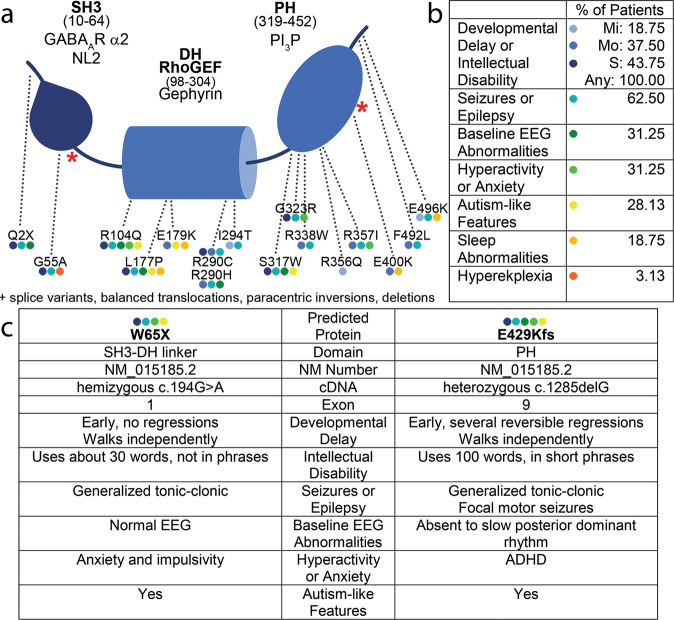
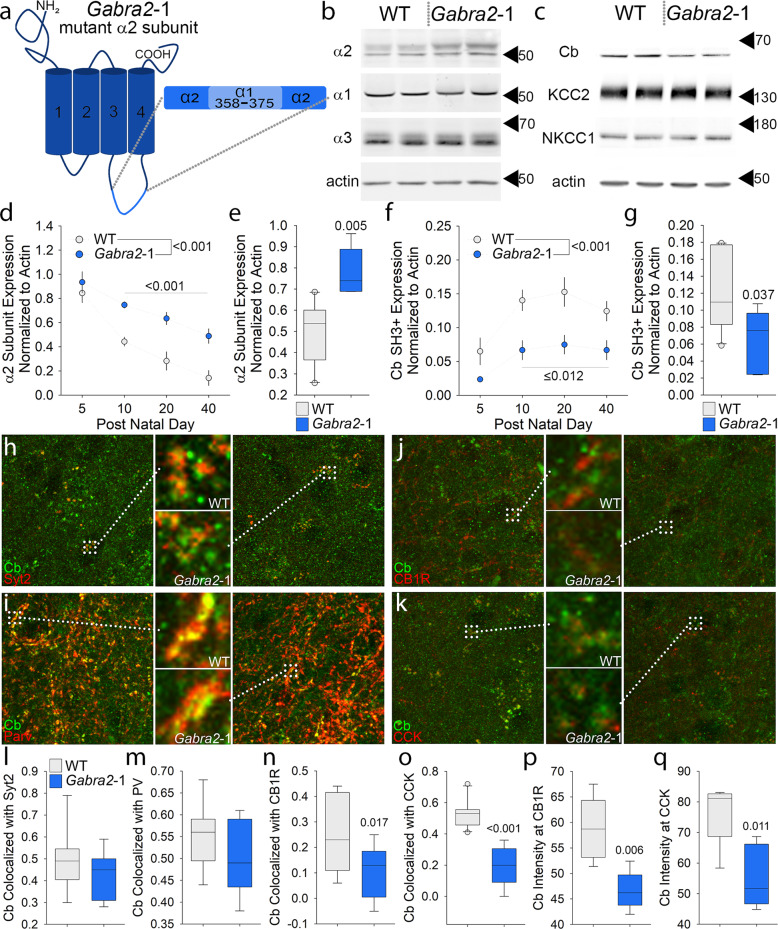
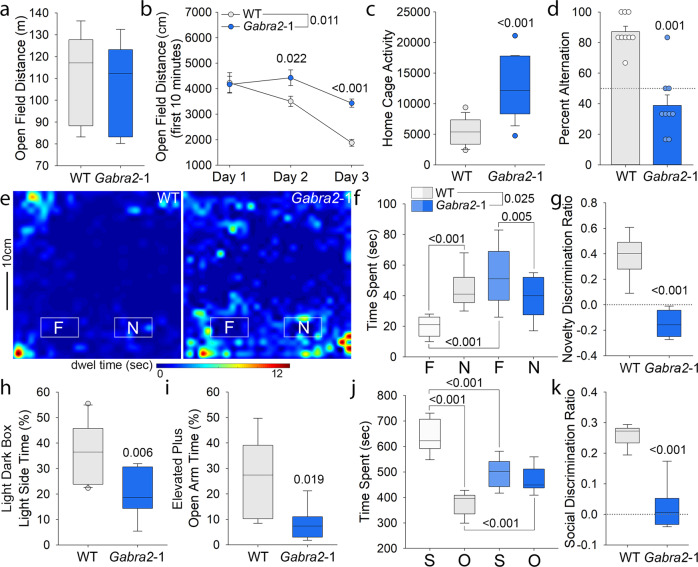
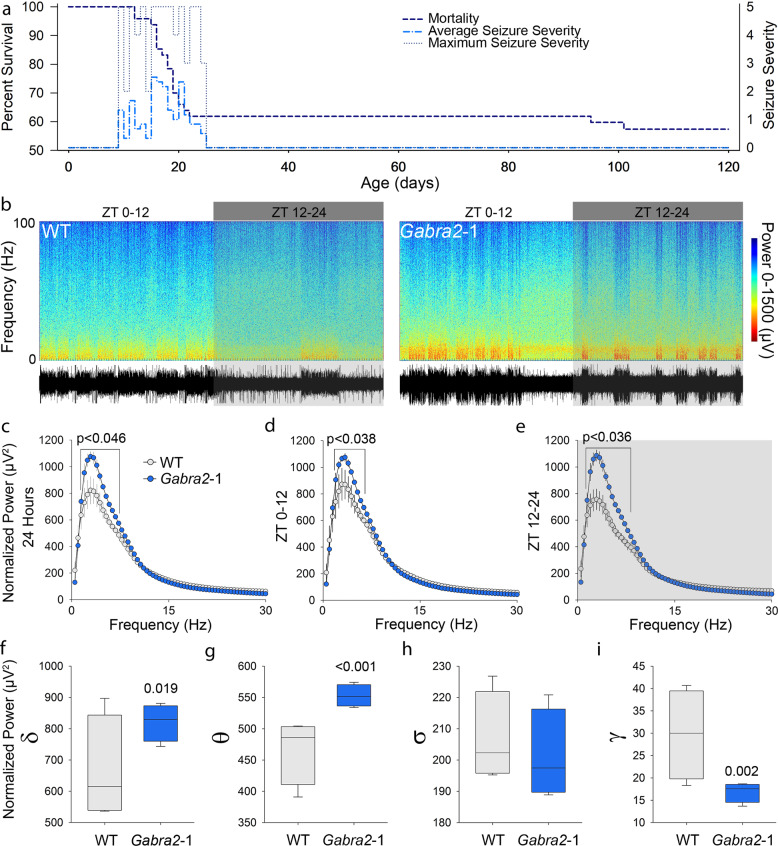
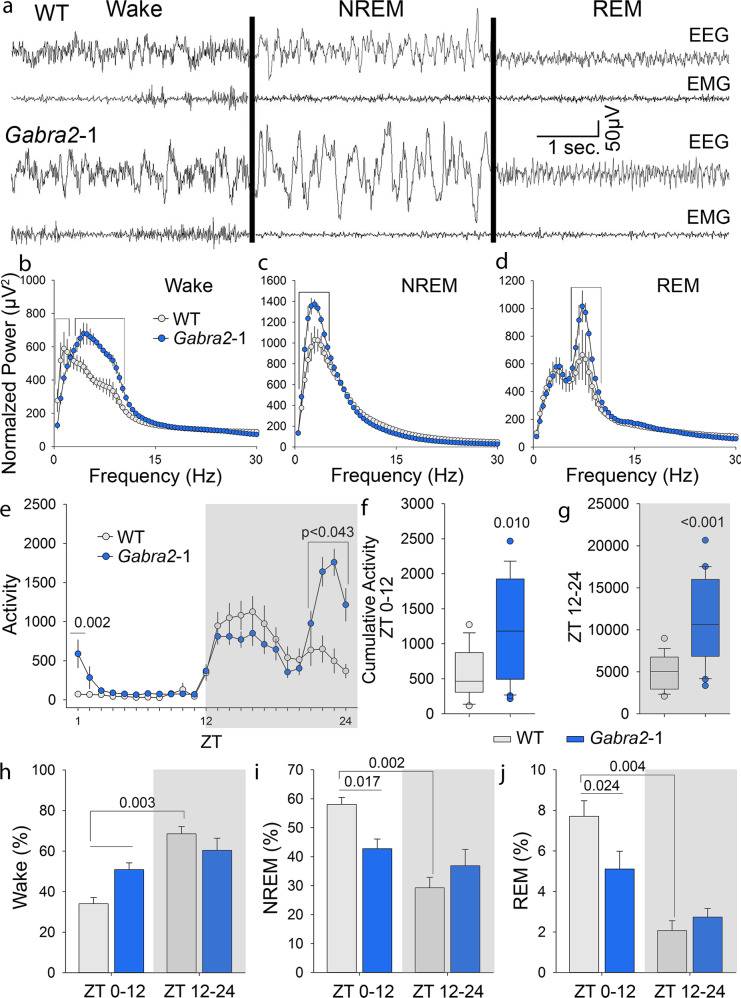
Intellectual disability (ID) is a common neurodevelopmental disorder that can arise from genetic mutations ranging from trisomy to single nucleotide polymorphism. Mutations in a growing number of single genes have been identified as causative in ID, including ARHGEF9. Evaluation of 41 ARHGEF9 patient reports shows ubiquitous inclusion of ID, along with other frequently reported symptoms of epilepsy, abnormal baseline EEG activity, behavioral symptoms, and sleep disturbances. ARHGEF9 codes for the Cdc42 Guanine Nucleotide Exchange Factor 9 collybistin (Cb), a known regulator of inhibitory synapse function via direct interaction with the adhesion molecule neuroligin-2 and the α2 subunit of GABAA receptors. We mutate the Cb binding motif within the large intracellular loop of α2 replacing it with the binding motif for gephyrin from the α1 subunit (Gabra2-1). The Gabra2-1 mutation causes a strong downregulation of Cb expression, particularly at cholecystokinin basket cell inhibitory synapses. Gabra2-1 mice have deficits in working and recognition memory, as well as hyperactivity, anxiety, and reduced social preference, recapitulating the frequently reported features of ARHGEF9 patients. Gabra2-1 mice also have spontaneous seizures during postnatal development which can lead to mortality, and baseline abnormalities in low-frequency wavelengths of the EEG. EEG abnormalities are vigilance state-specific and manifest as sleep disturbance including increased time in wake and a loss of free-running rhythmicity in the absence of light as zeitgeber. Gabra2-1 mice phenocopy multiple features of human ARHGEF9 mutation, and reveal α2 subunit-containing GABAA receptors as a druggable target for treatment of this complex ID syndrome.
© 2022. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: SJM serves as a consultant for AstraZeneca, Bain Capital, and SAGE Therapeutics, relationships that are regulated by Tufts University. SJM is also a shareholder in SAGE Therapeutics.
Figures
References
-
- de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. 101056/NEJMoa1206524. 2012. 10.1056/NEJMoa1206524. Accessed 21 April 2021. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
