

Champagne: Automated Whole-Genome Phylogenomic Character Matrix Method Using Large Genomic Indels for Homoplasy-Free Inference
- PMID: 35171243
- PMCID: PMC8920512
- DOI: 10.1093/gbe/evac013
Champagne: Automated Whole-Genome Phylogenomic Character Matrix Method Using Large Genomic Indels for Homoplasy-Free Inference
Abstract
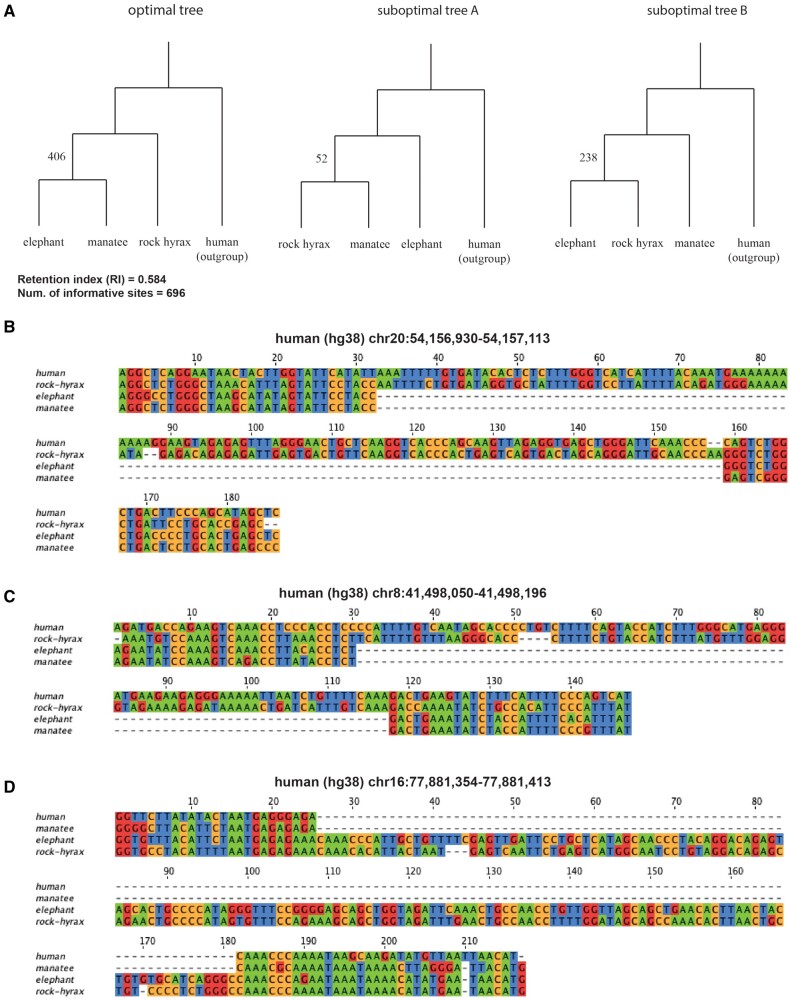
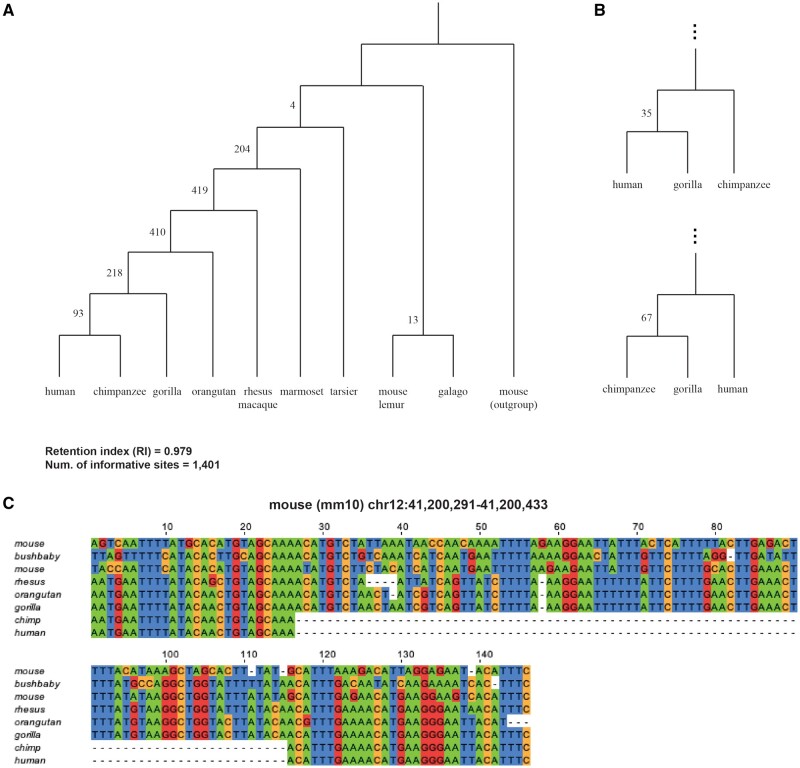
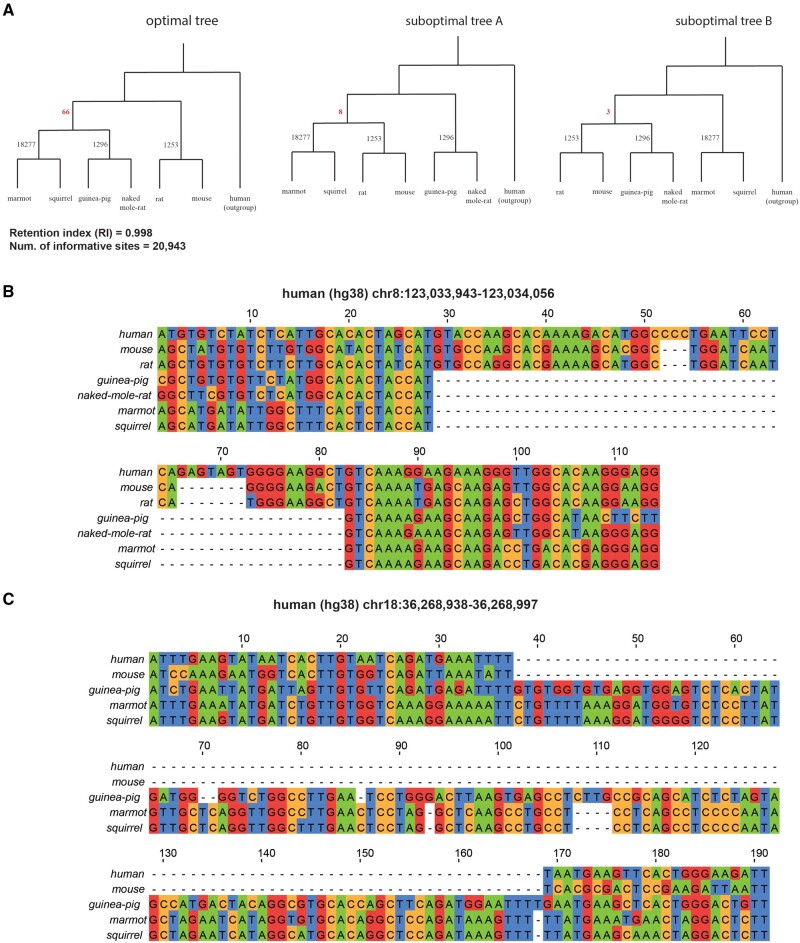
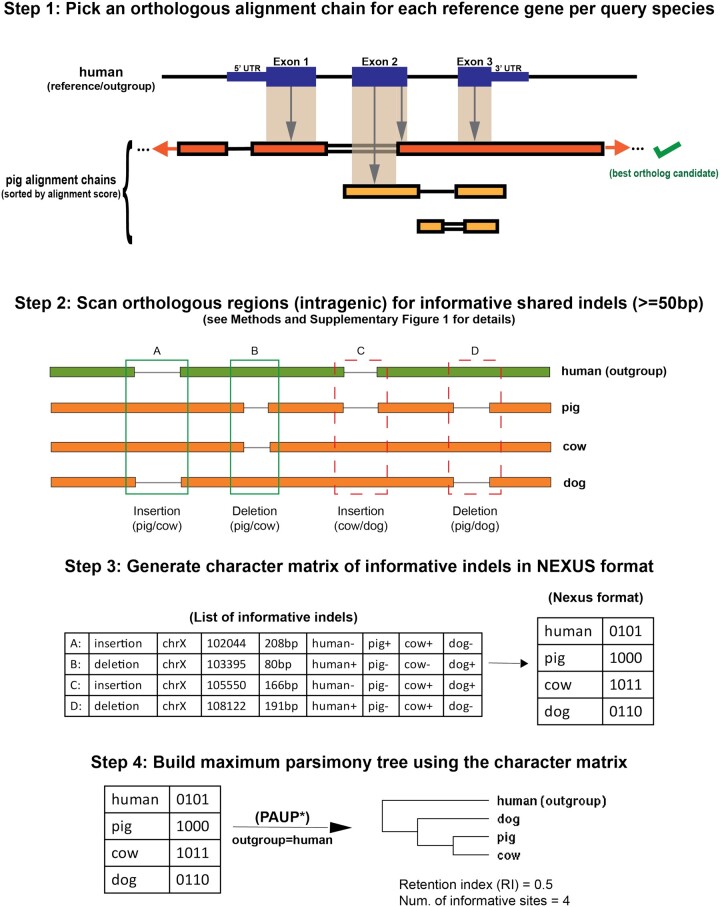
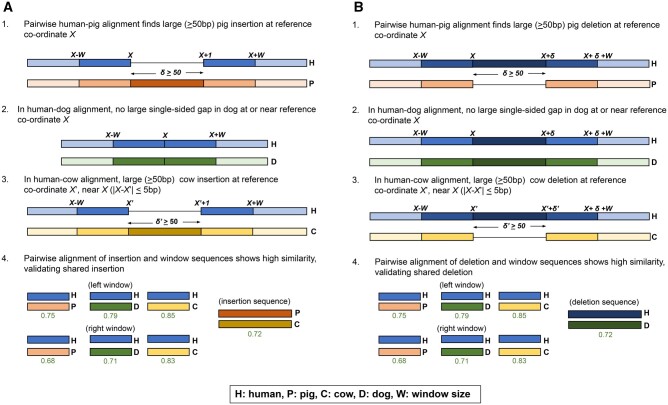
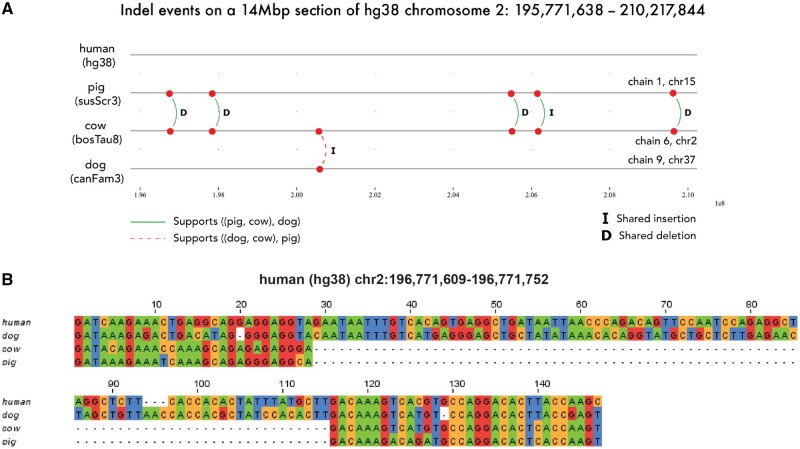
We present Champagne, a whole-genome method for generating character matrices for phylogenomic analysis using large genomic indel events. By rigorously picking orthologous genes and locating large insertion and deletion events, Champagne delivers a character matrix that considerably reduces homoplasy compared with morphological and nucleotide-based matrices, on both established phylogenies and difficult-to-resolve nodes in the mammalian tree. Champagne provides ample evidence in the form of genomic structural variation to support incomplete lineage sorting and possible introgression in Paenungulata and human-chimp-gorilla which were previously inferred primarily through matrices composed of aligned single-nucleotide characters. Champagne also offers further evidence for Myomorpha as sister to Sciuridae and Hystricomorpha in the rodent tree. Champagne harbors distinct theoretical advantages as an automated method that produces nearly homoplasy-free character matrices on the whole-genome scale.
Keywords: homoplasy-free characters; incomplete lineage sorting; phylogenetics; phylogenomics; rare genomic changes.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Bejerano G, et al.2004. Ultraconserved elements in the human genome. Science 304(5675):1321–1325. - PubMed
-
- Belyayev A. 2014. Bursts of transposable elements as an evolutionary driving force. J Evol Biol. 27(12):2573–2584. - PubMed
-
- Bergsten J. 2005. A review of long-branch attraction. Cladistics 21(2):163–193. - PubMed
