

Propofol toxicity in the developing mouse heart mitochondria
- PMID: 35173299
- PMCID: PMC9378757
- DOI: 10.1038/s41390-022-01985-1
Propofol toxicity in the developing mouse heart mitochondria
Abstract
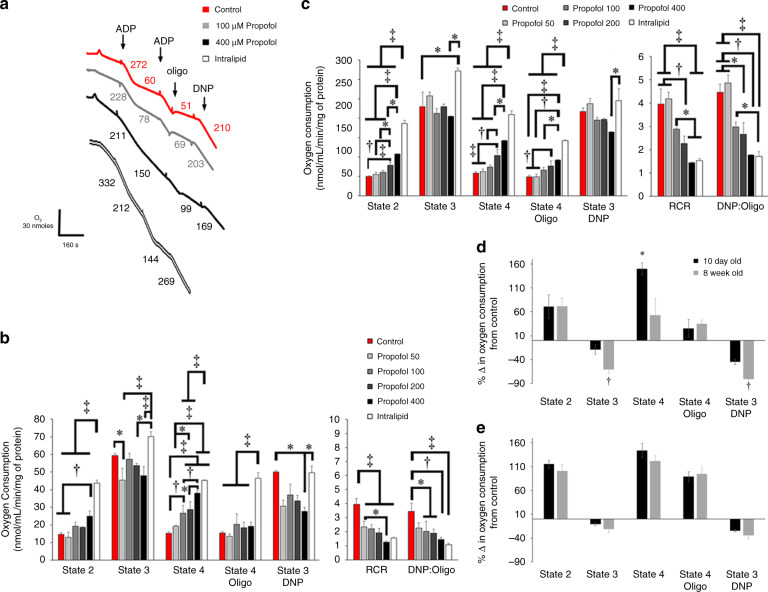
Background: Propofol infusion syndrome (PRIS) is a potentially lethal consequence of long-term propofol administration. Children are vulnerable and cardiac involvement is often prominent and associated with mortality. We aimed to determine the mechanism of propofol toxicity in newborn mice, hypothesizing that propofol would induce discrete defects within immature cardiac mitochondria.
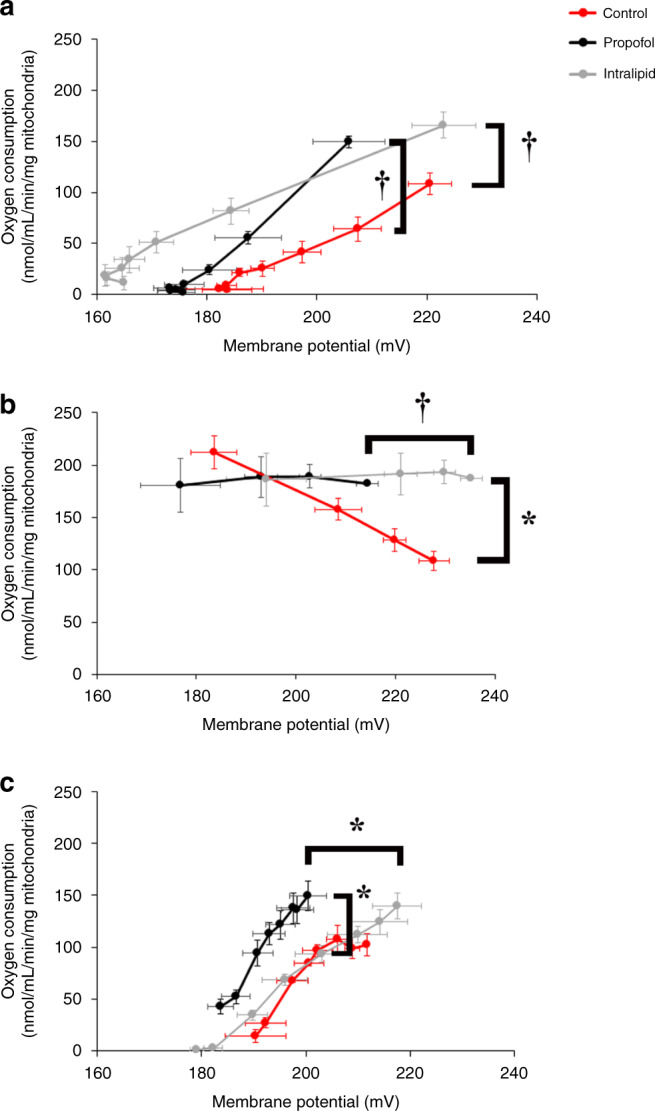
Methods: Newborn murine cardiac mitochondria were exposed to propofol or intralipid in vitro. Non-exposed mitochondria served as controls. Mitochondrial respiration and membrane potential (ΔΨ) were measured and respiratory chain complex kinetics were determined.
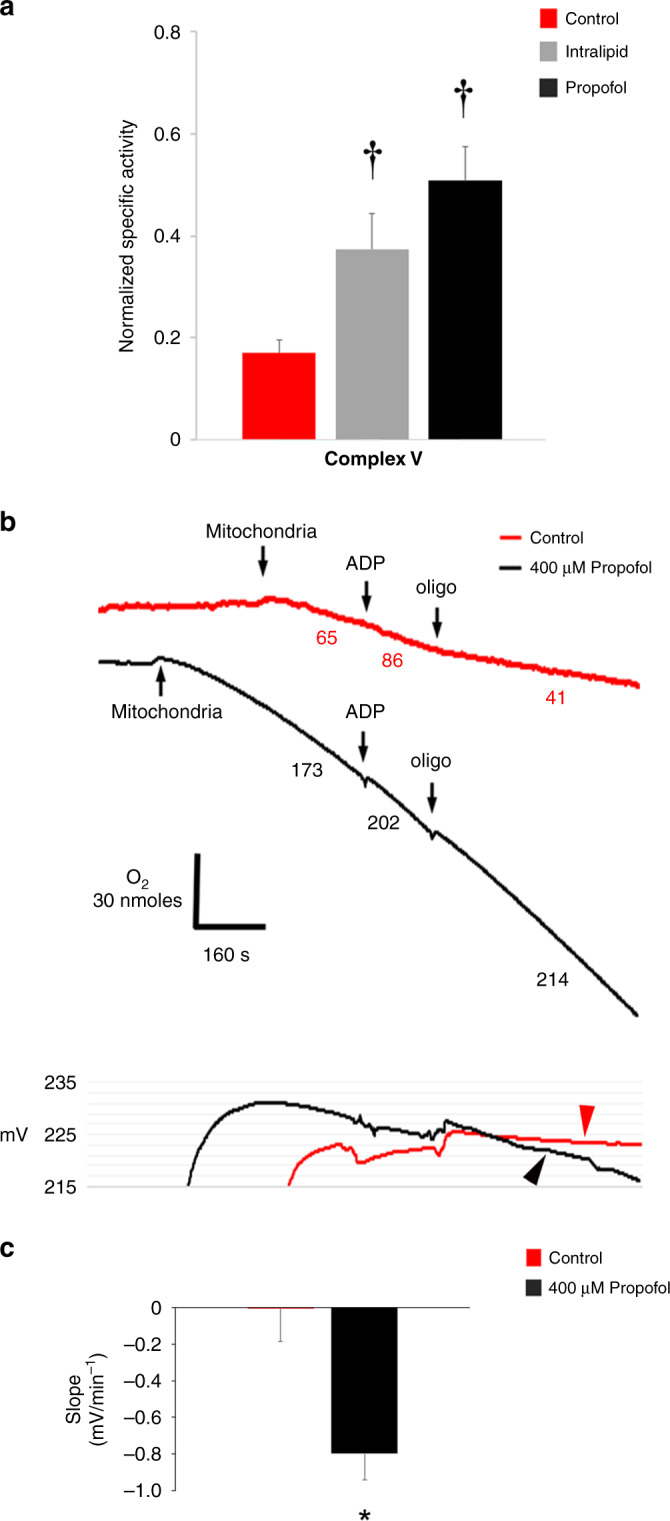
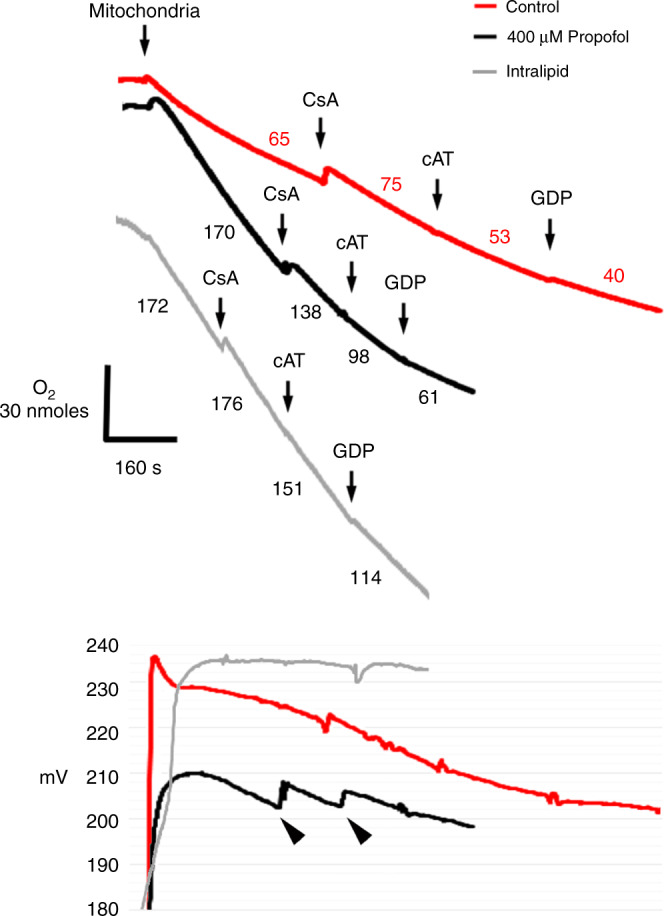
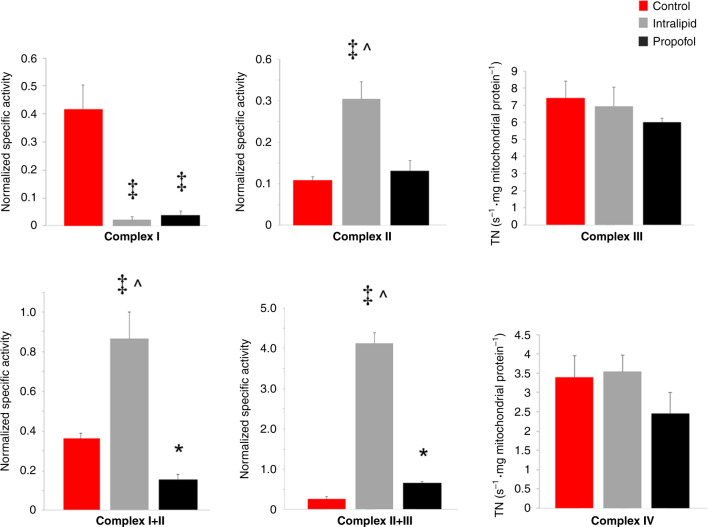
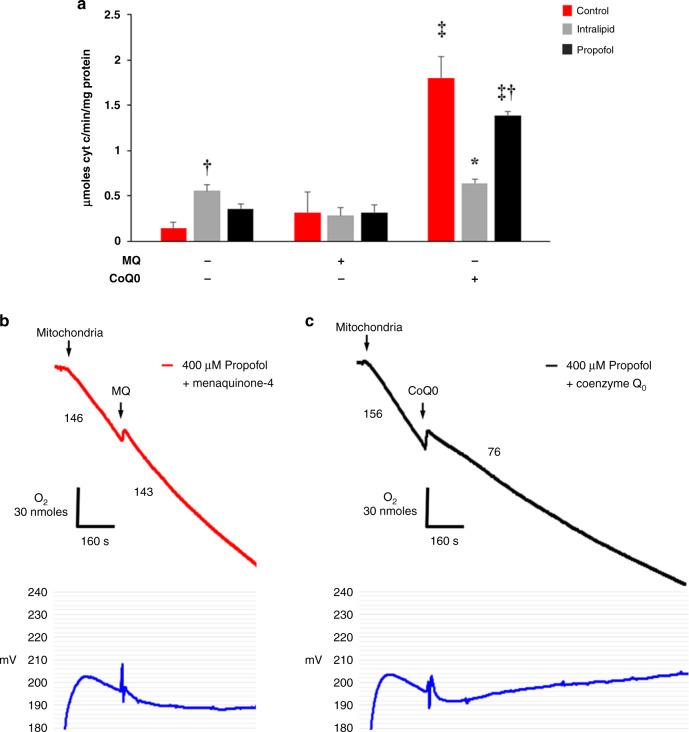
Results: Propofol and intralipid exerted biological activity in isolated mitochondria. Although intralipid effects were a potential confounder, we found that propofol induced a dose-dependent increase in proton leak and caused a defect in substrate oxidation at coenzyme Q (CoQ). These impairments prevented propofol-exposed cardiomyocyte mitochondria from generating an adequate ΔΨ. The addition of the quinone analog, CoQ0, blocked propofol-induced leak and increased Complex II+III activity.
Conclusions: Propofol uncoupled immature cardiomyocyte mitochondria by inducing excessive CoQ-sensitive leak and interfered with electron transport at CoQ. The findings provide new insight into the mechanisms of propofol toxicity in the developing heart and may help explain why children are vulnerable to developing PRIS.
Impact: Propofol uncouples immature cardiomyocyte mitochondria by inducing excessive coenzyme Q (CoQ)-sensitive proton leak. Propofol also interferes with electron transport at the level of CoQ. These defects provide new insight into propofol toxicity in the developing heart.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
