

Challenges for Clinical Drug Development in Pulmonary Fibrosis
- PMID: 35173620
- PMCID: PMC8841605
- DOI: 10.3389/fphar.2022.823085
Challenges for Clinical Drug Development in Pulmonary Fibrosis
Abstract
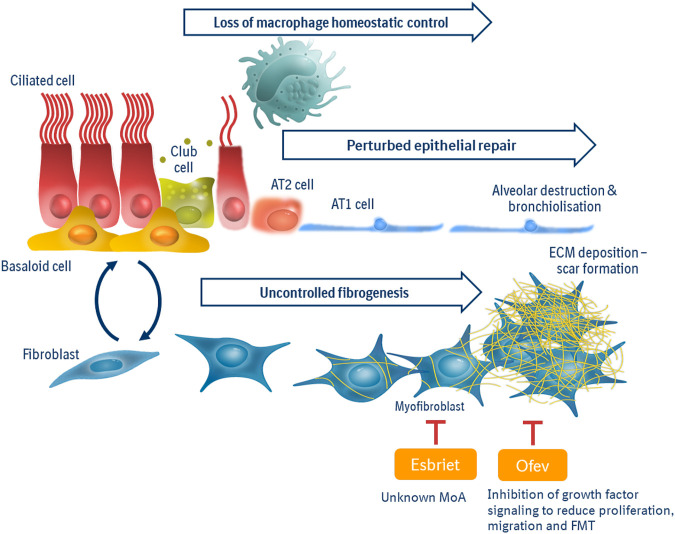
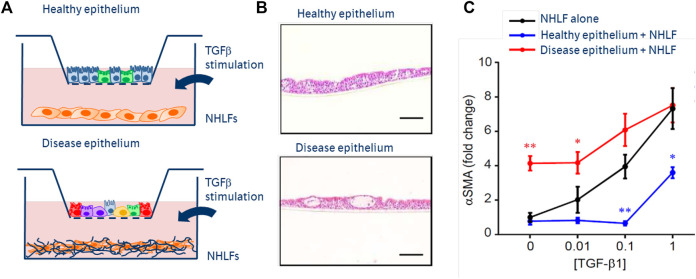
Pulmonary fibrosis is a pathologic process associated with scarring of the lung interstitium. Interstitial lung diseases (ILDs) encompass a large and heterogenous group of disorders, a number of which are characterized by progressive pulmonary fibrosis that leads to respiratory failure and death. Idiopathic pulmonary fibrosis (IPF) has been described as an archetype of progressive fibrosing ILD, and the development of pirfenidone and nintedanib has been a major breakthrough in the treatment of patients with this deadly disease. Both drugs principally target scar-forming fibroblasts and have been shown to significantly slow down the accelerated decline of lung function by approximately 50%. In addition, nintedanib has been approved for patients with other progressive fibrosing ILDs and systemic sclerosis-associated ILD. However, there is still no cure for pulmonary fibrosis and no meaningful improvement of symptoms or quality of life has been shown. Advancement in research, such as the advent of single cell sequencing technology, has identified additional pathologic cell populations beyond the fibroblast which could be targeted for therapeutic purposes. The preclinical and clinical development of novel drug candidates is hampered by profound challenges such as a lack of sensitive clinical outcomes or suitable biomarkers that would provide an early indication of patient benefit. With the availability of these anti-fibrotic treatments, it has become even more difficult to demonstrate added efficacy, in particular in short-term clinical studies. Patient heterogeneity and the paucity of biomarkers of disease activity further complicate clinical development. It is conceivable that future treatment of pulmonary fibrosis will need to embrace more precision in treating the right patient at the right time, explore novel measures of efficacy, and likely combine treatment options.
Keywords: clinical trial; drugs; fibroblast; interstitial; lung disease; pharmaceutical research; pharmacology.
Copyright © 2022 White, Thomas, Stowasser and Tetzlaff.
Conflict of interest statement
Author EW is employed by the company Boehringer Ingelheim Pharmaceuticals, Inc. Author MT is employed by the company Boehringer Ingelheim Pharma GmbH & Co. KG. Authors SS and KT are employed by the company Boehringer Ingelheim International GmbH. The page processing charges for this article have been paid by Boehringer Ingelheim.
Figures
Similar articles
-
Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases.Eur Respir J. 2019 Sep 19;54(3):1900161. doi: 10.1183/13993003.00161-2019. Print 2019 Sep. Eur Respir J. 2019. PMID: 31285305 Free PMC article. Review.
-
Progressive fibrosing interstitial lung diseases: A new concept and indication of nintedanib.Mod Rheumatol. 2021 Jan;31(1):13-19. doi: 10.1080/14397595.2020.1826665. Mod Rheumatol. 2021. PMID: 32964766 Review.
-
Possible value of antifibrotic drugs in patients with progressive fibrosing non-IPF interstitial lung diseases.BMC Pulm Med. 2019 Nov 12;19(1):213. doi: 10.1186/s12890-019-0937-0. BMC Pulm Med. 2019. PMID: 31718637 Free PMC article.
-
Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial.Lancet Respir Med. 2021 May;9(5):476-486. doi: 10.1016/S2213-2600(20)30554-3. Epub 2021 Mar 30. Lancet Respir Med. 2021. PMID: 33798455 Clinical Trial.
-
Progressive Fibrosing Interstitial Lung Diseases: A Current Perspective.Biomedicines. 2021 Sep 16;9(9):1237. doi: 10.3390/biomedicines9091237. Biomedicines. 2021. PMID: 34572422 Free PMC article. Review.
Cited by
-
Dinebra retroflexa Herbal Phytotherapy: A Simulation Study Based on Bleomycin-Induced Pulmonary Fibrosis Retraction Potential in Swiss Albino Rats.Medicina (Kaunas). 2022 Nov 24;58(12):1719. doi: 10.3390/medicina58121719. Medicina (Kaunas). 2022. PMID: 36556921 Free PMC article.
-
PGF2α signaling drives fibrotic remodeling and fibroblast population dynamics in mice.JCI Insight. 2023 Dec 22;8(24):e172977. doi: 10.1172/jci.insight.172977. JCI Insight. 2023. PMID: 37934604 Free PMC article.
-
Disruption of Prostaglandin F2α Receptor Signaling Attenuates Fibrotic Remodeling and Alters Fibroblast Population Dynamics in A Preclinical Murine Model of Idiopathic Pulmonary Fibrosis.bioRxiv [Preprint]. 2023 Jun 7:2023.06.07.543956. doi: 10.1101/2023.06.07.543956. bioRxiv. 2023. Update in: JCI Insight. 2023 Dec 22;8(24):e172977. doi: 10.1172/jci.insight.172977. PMID: 37333249 Free PMC article. Updated. Preprint.
-
Targeting pathogenic macrophages by the application of SHP-1 agonists reduces inflammation and alleviates pulmonary fibrosis.Cell Death Dis. 2023 Jun 8;14(6):352. doi: 10.1038/s41419-023-05876-z. Cell Death Dis. 2023. PMID: 37291088 Free PMC article.
-
Biodistribution, Dosimetry, and Pharmacokinetics of 68Ga-CBP8: A Type I Collagen-Targeted PET Probe.J Nucl Med. 2023 May;64(5):775-781. doi: 10.2967/jnumed.122.264530. Epub 2022 Dec 8. J Nucl Med. 2023. PMID: 37116909 Free PMC article.
References
-
- Behr J., Prasse A., Kreuter M., Johow J., Rabe K. F., Bonella F., et al. (2021). Pirfenidone in Patients with Progressive Fibrotic Interstitial Lung Diseases Other Than Idiopathic Pulmonary Fibrosis (RELIEF): A Double-Blind, Randomised, Placebo-Controlled, Phase 2b Trial. Lancet Respir. Med. 9, 476–486. 10.1016/s2213-2600(20)30554-3 - DOI - PubMed
-
- Behr J., Prasse A., Wirtz H., Koschel D., Pittrow D., Held M., et al. (2020). Survival and Course of Lung Function in the Presence or Absence of Antifibrotic Treatment in Patients with Idiopathic Pulmonary Fibrosis: Long-Term Results of the INSIGHTS-IPF Registry. Eur. Respir. J. 56, 1902279. 10.1183/13993003.02279-2019 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources