

Integrated Genomic Analysis Identifies UBTF Tandem Duplications as a Recurrent Lesion in Pediatric Acute Myeloid Leukemia
- PMID: 35176137
- PMCID: PMC9780084
- DOI: 10.1158/2643-3230.BCD-21-0160
Integrated Genomic Analysis Identifies UBTF Tandem Duplications as a Recurrent Lesion in Pediatric Acute Myeloid Leukemia
Abstract
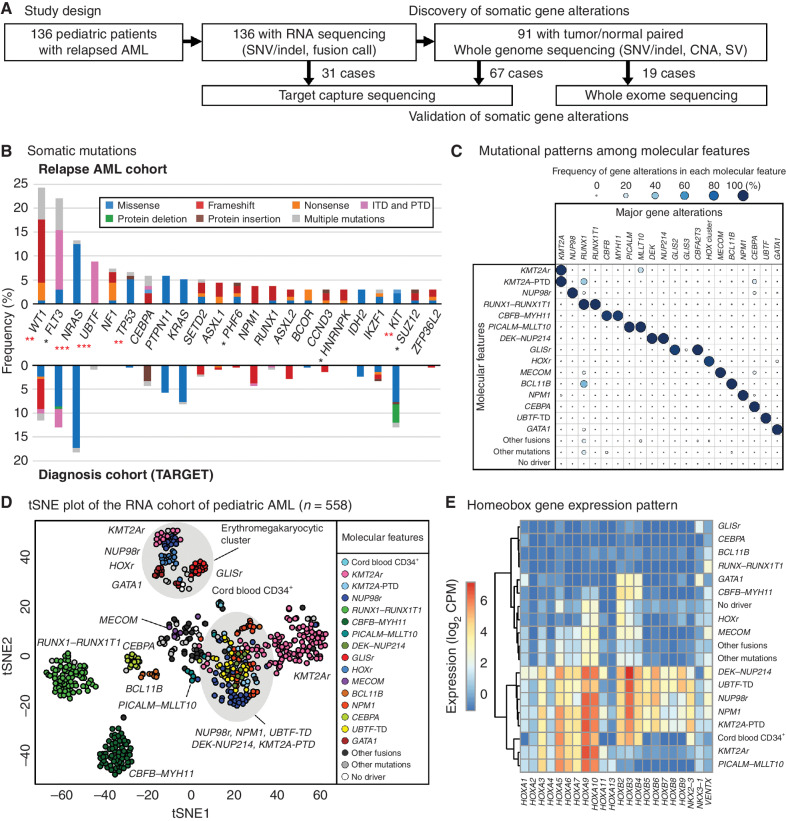
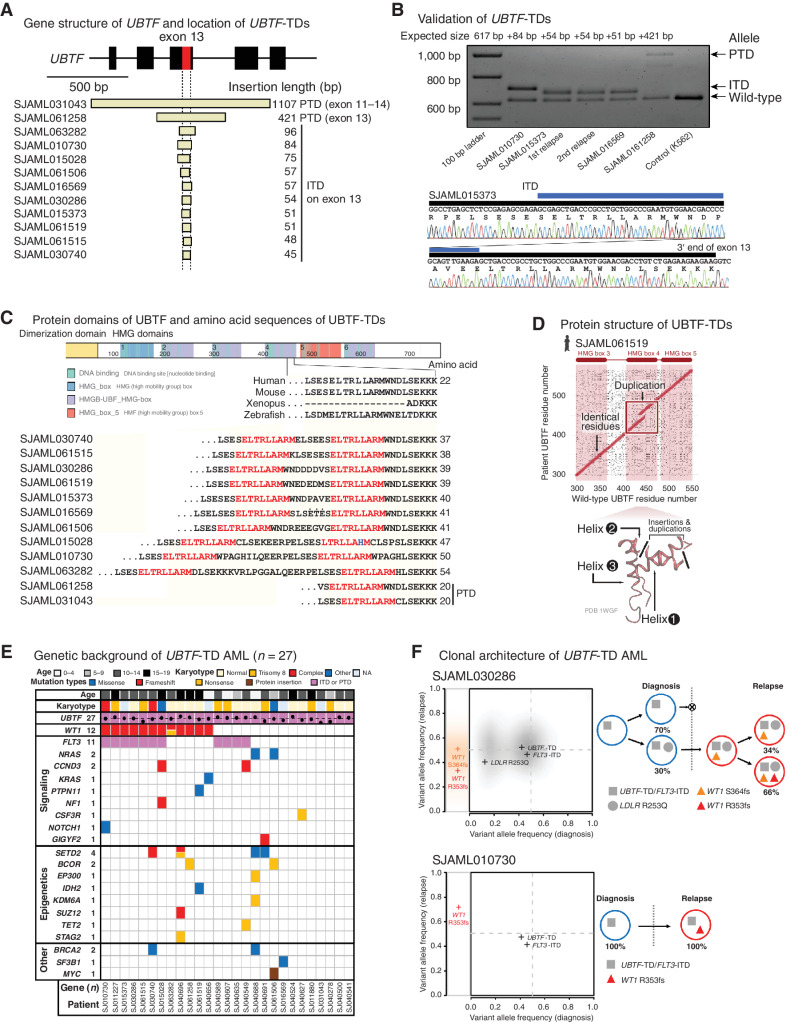
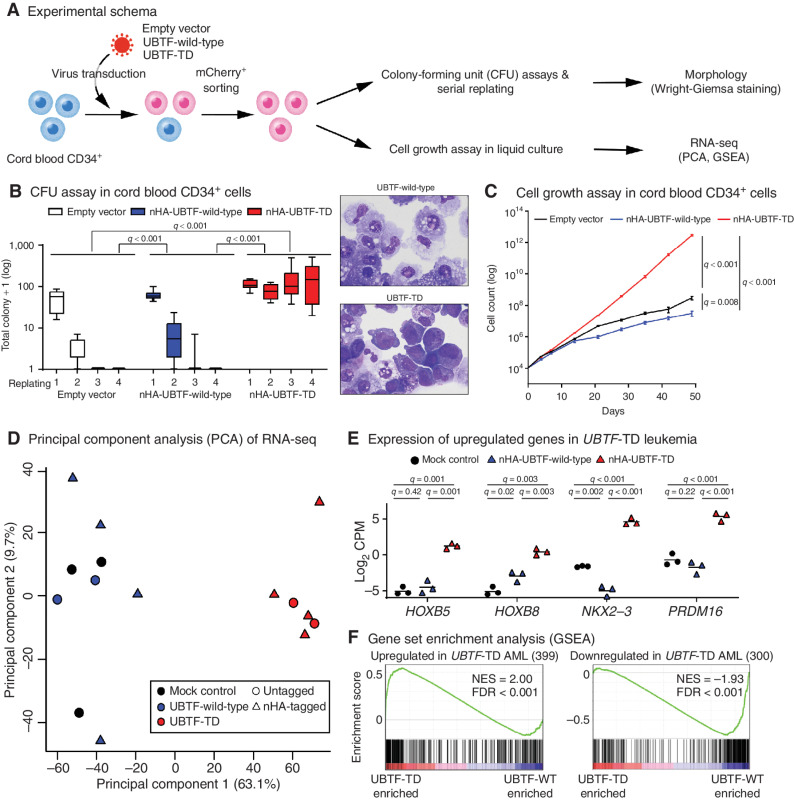
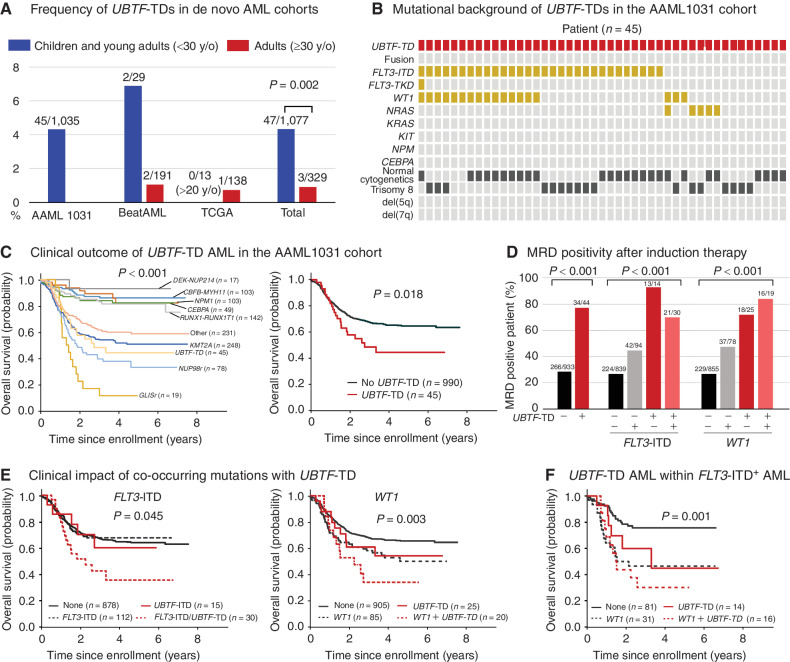
The genetics of relapsed pediatric acute myeloid leukemia (AML) has yet to be comprehensively defined. Here, we present the spectrum of genomic alterations in 136 relapsed pediatric AMLs. We identified recurrent exon 13 tandem duplications (TD) in upstream binding transcription factor (UBTF) in 9% of relapsed AML cases. UBTF-TD AMLs commonly have normal karyotype or trisomy 8 with cooccurring WT1 mutations or FLT3-ITD but not other known oncogenic fusions. These UBTF-TD events are stable during disease progression and are present in the founding clone. In addition, we observed that UBTF-TD AMLs account for approximately 4% of all de novo pediatric AMLs, are less common in adults, and are associated with poor outcomes and MRD positivity. Expression of UBTF-TD in primary hematopoietic cells is sufficient to enhance serial clonogenic activity and to drive a similar transcriptional program to UBTF-TD AMLs. Collectively, these clinical, genomic, and functional data establish UBTF-TD as a new recurrent mutation in AML.
Significance: We defined the spectrum of mutations in relapsed pediatric AML and identified UBTF-TDs as a new recurrent genetic alteration. These duplications are more common in children and define a group of AMLs with intermediate-risk cytogenetic abnormalities, FLT3-ITD and WT1 alterations, and are associated with poor outcomes. See related commentary by Hasserjian and Nardi, p. 173. This article is highlighted in the In This Issue feature, p. 171.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
-
Bedside to Bench and Back: Identifying a New Clinically Relevant Driver in Pediatric Acute Myeloid Leukemia.Blood Cancer Discov. 2022 May 5;3(3):173-175. doi: 10.1158/2643-3230.BCD-22-0004. Blood Cancer Discov. 2022. PMID: 35176140 Free PMC article.
Comment on
-
Bedside to Bench and Back: Identifying a New Clinically Relevant Driver in Pediatric Acute Myeloid Leukemia.Blood Cancer Discov. 2022 May 5;3(3):173-175. doi: 10.1158/2643-3230.BCD-22-0004. Blood Cancer Discov. 2022. PMID: 35176140 Free PMC article.
References
-
- Cornelissen JJ, Gratwohl A, Schlenk RF, Sierra J, Bornhauser M, Juliusson Get al. The European LeukemiaNet AML Working Party consensus statement on allogeneic HSCT for patients with AML in remission: an integrated-risk adapted approach. Nat Rev Clin Oncol 2012;9:579–90. - PubMed
-
- Kaspers GJ, Zimmermann M, Reinhardt D, Gibson BE, Tamminga RY, Aleinikova Oet al. Improved outcome in pediatric relapsed acute myeloid leukemia: results of a randomized trial on liposomal daunorubicin by the International BFM Study Group. J Clin Oncol 2013;31:599–607. - PubMed
-
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, Pratcorona M, Abbas S, Kuipers JEet al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011;118:3645–56. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
