

A cattle graph genome incorporating global breed diversity
- PMID: 35177600
- PMCID: PMC8854726
- DOI: 10.1038/s41467-022-28605-0
A cattle graph genome incorporating global breed diversity
Erratum in
-
Author Correction: A cattle graph genome incorporating global breed diversity.Nat Commun. 2022 May 23;13(1):2983. doi: 10.1038/s41467-022-30372-x. Nat Commun. 2022. PMID: 35606359 Free PMC article. No abstract available.
Abstract
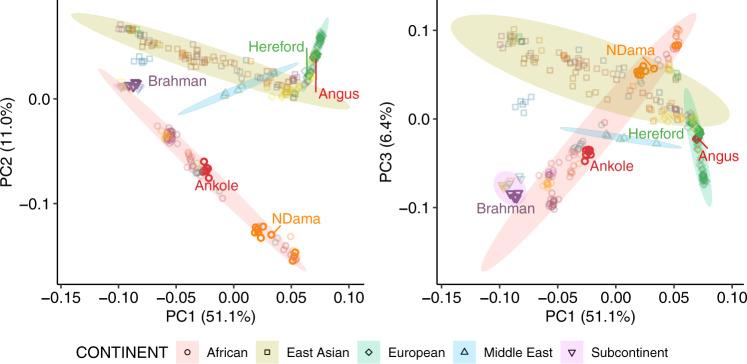
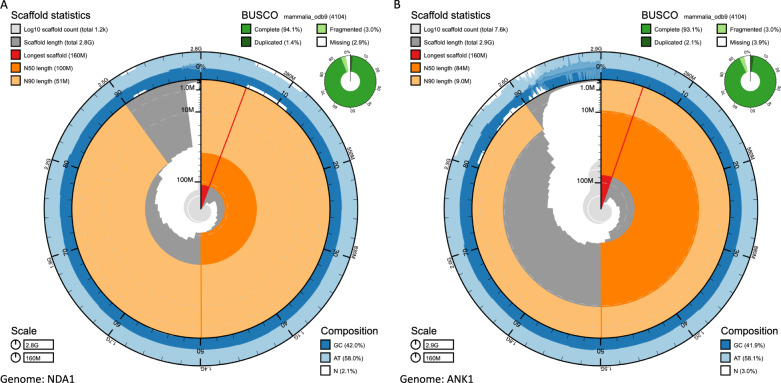
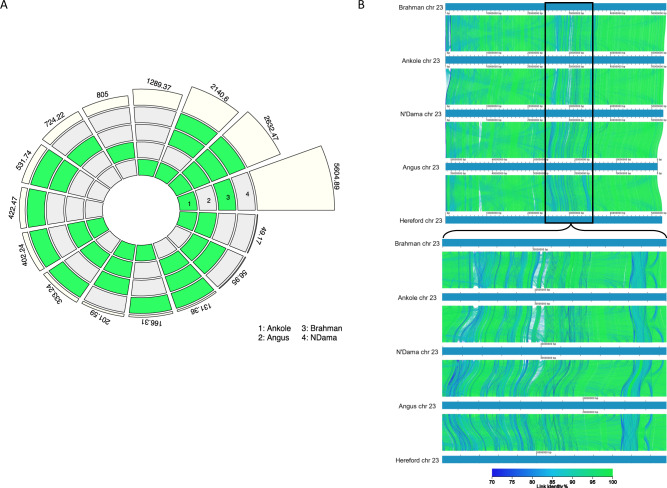
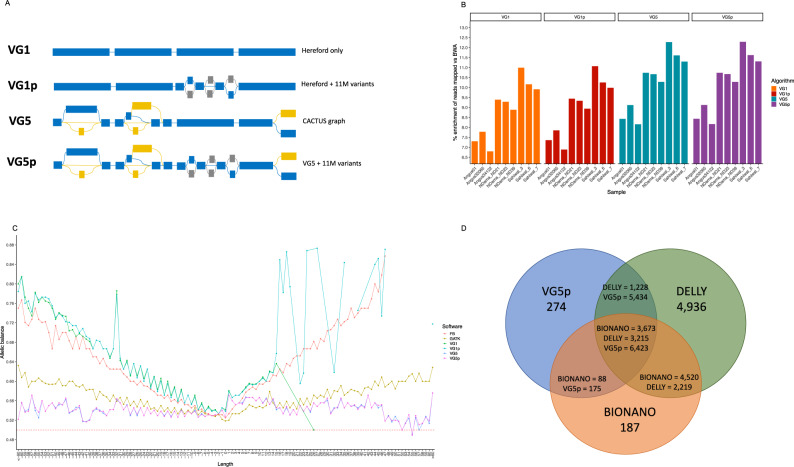
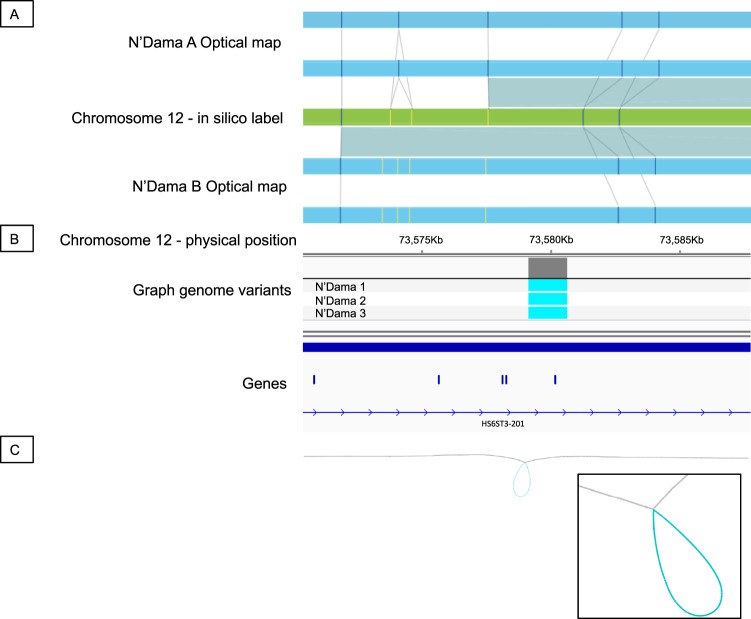
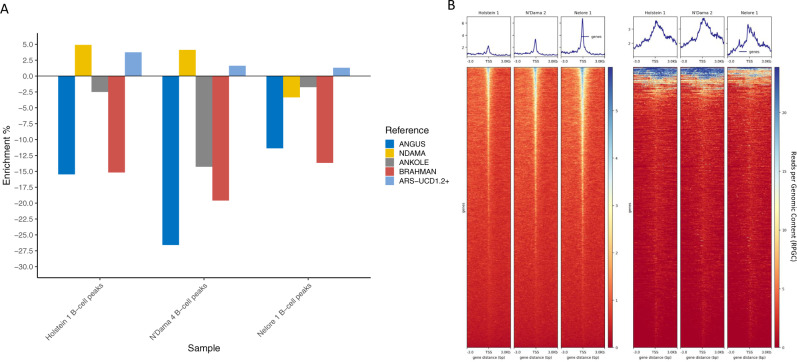
Despite only 8% of cattle being found in Europe, European breeds dominate current genetic resources. This adversely impacts cattle research in other important global cattle breeds, especially those from Africa for which genomic resources are particularly limited, despite their disproportionate importance to the continent's economies. To mitigate this issue, we have generated assemblies of African breeds, which have been integrated with genomic data for 294 diverse cattle into a graph genome that incorporates global cattle diversity. We illustrate how this more representative reference assembly contains an extra 116.1 Mb (4.2%) of sequence absent from the current Hereford sequence and consequently inaccessible to current studies. We further demonstrate how using this graph genome increases read mapping rates, reduces allelic biases and improves the agreement of structural variant calling with independent optical mapping data. Consequently, we present an improved, more representative, reference assembly that will improve global cattle research.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- De Boer H. Cattle genetic resources. Livest. Prod. Sci. 1991;29:256–258. doi: 10.1016/0301-6226(91)90072-X. - DOI
-
- Felius M, et al. On the breeds of cattle-Historic and current classifications. Diversity. 2011;3:660–692. doi: 10.3390/d3040660. - DOI
-
- Ajmone-Marsan P, Lenstra JA, Fernando Garcia J, The Globaldiv Consortium. On the origin of cattle: how aurochs became domestic and colonized the world Attenuation of the inflammatory phenomena in the transition period of dairy cows View project Climate Genomics for Farm Animal Adaptation View project. Evol. Anthropol. 2010;19:148–157. doi: 10.1002/evan.20267. - DOI
Publication types
MeSH terms
Grants and funding
- BB/T019468/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/D/10002070/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/R015155/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/P024025/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Miscellaneous
