

Prognostic tools and candidate drugs based on plasma proteomics of patients with severe COVID-19 complications
- PMID: 35177642
- PMCID: PMC8854716
- DOI: 10.1038/s41467-022-28639-4
Prognostic tools and candidate drugs based on plasma proteomics of patients with severe COVID-19 complications
Abstract
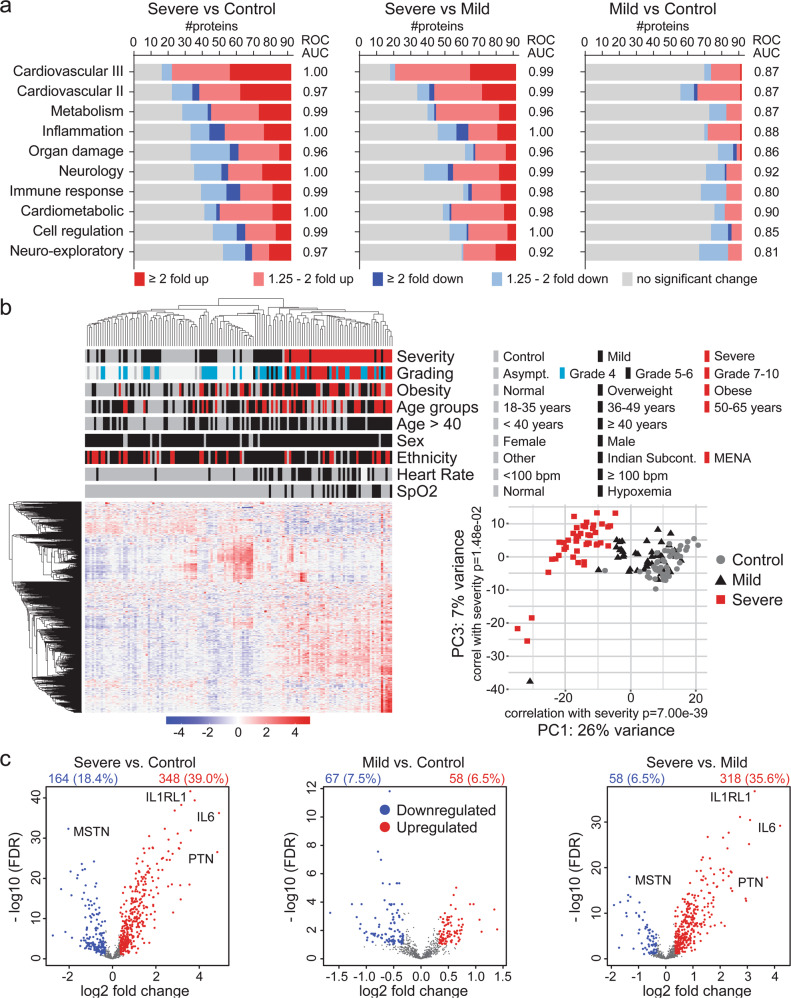
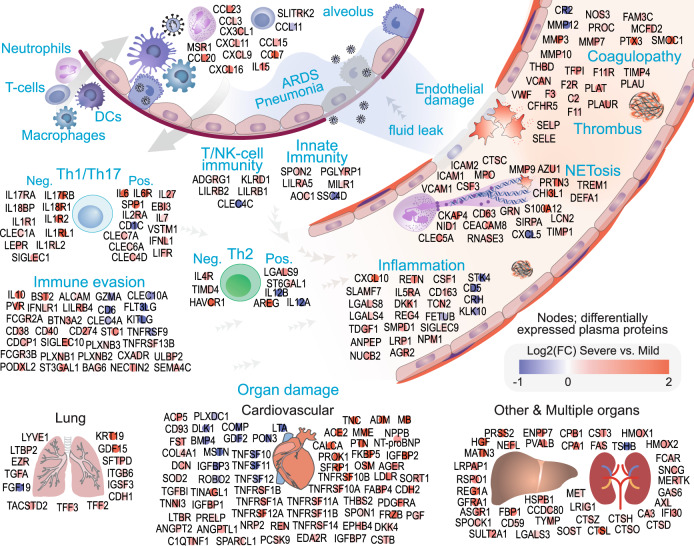
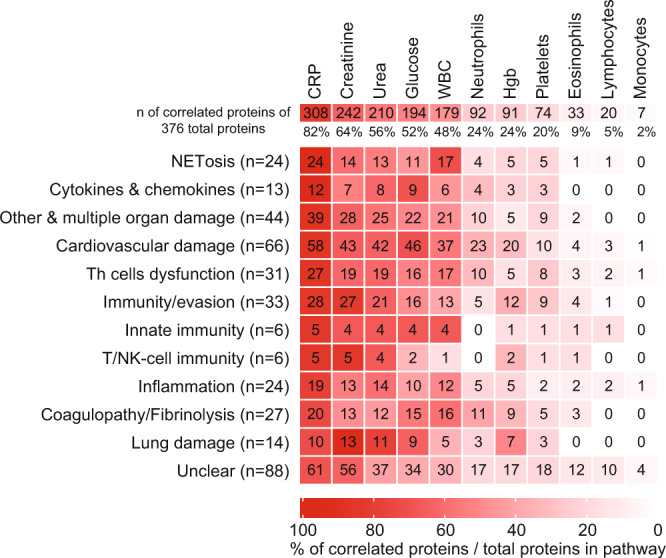
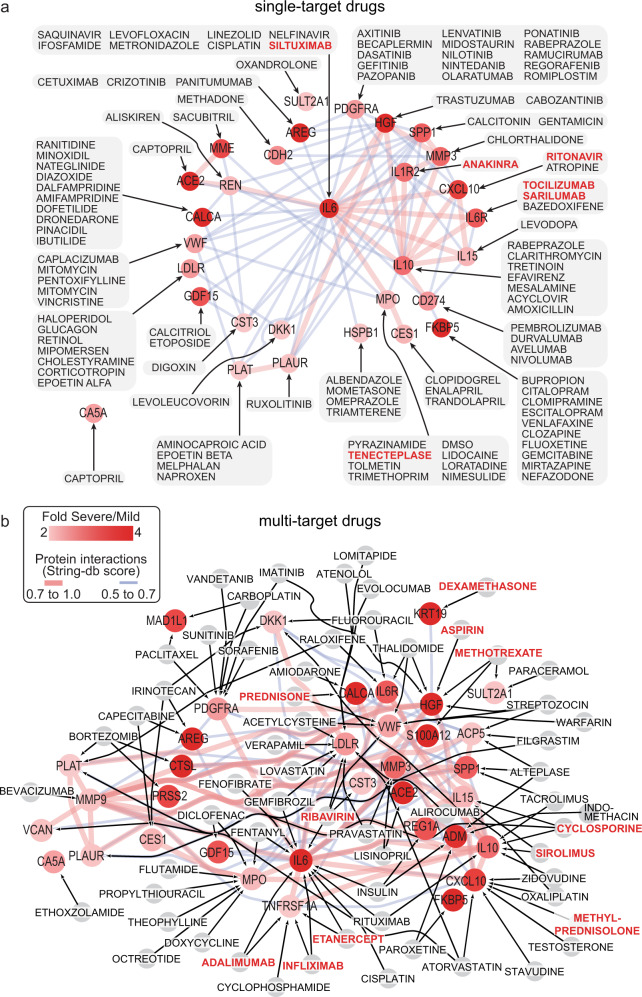
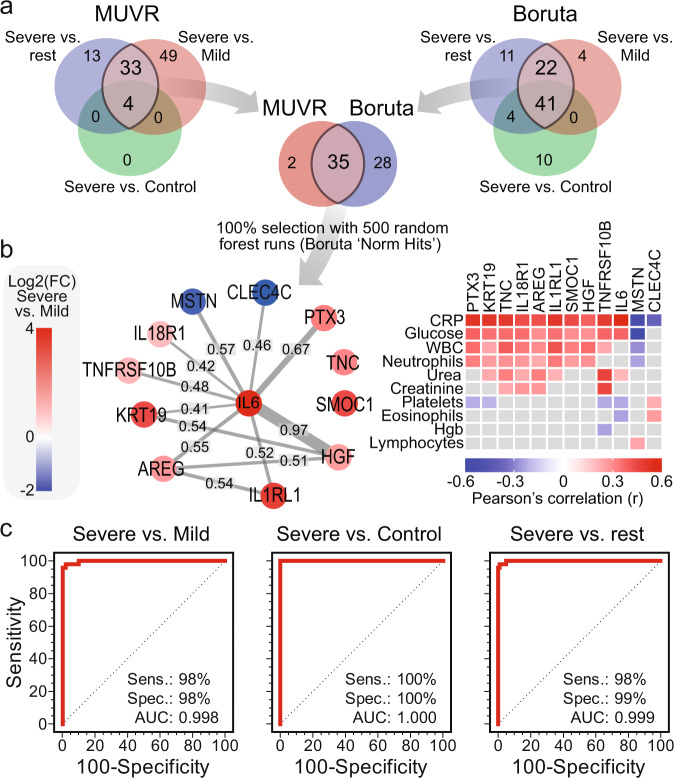
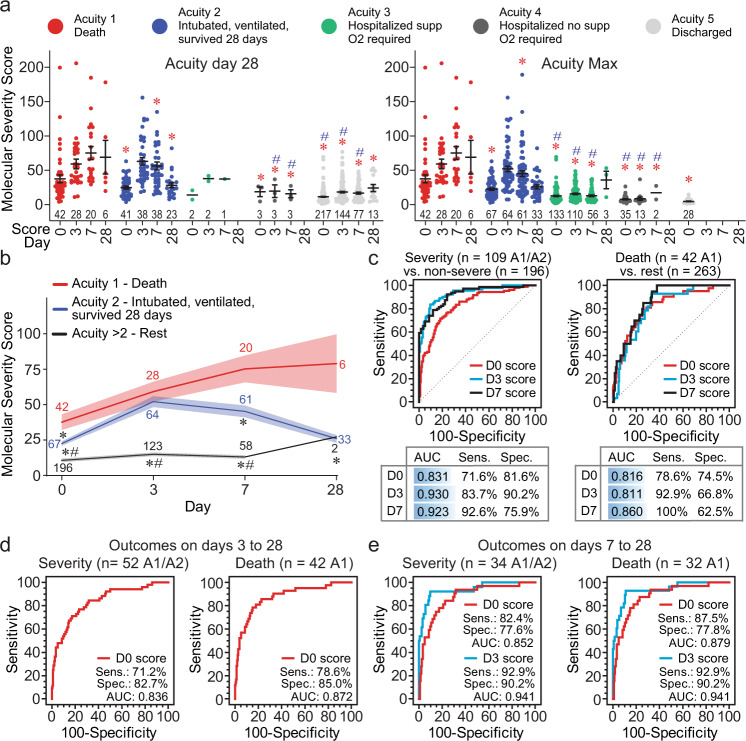
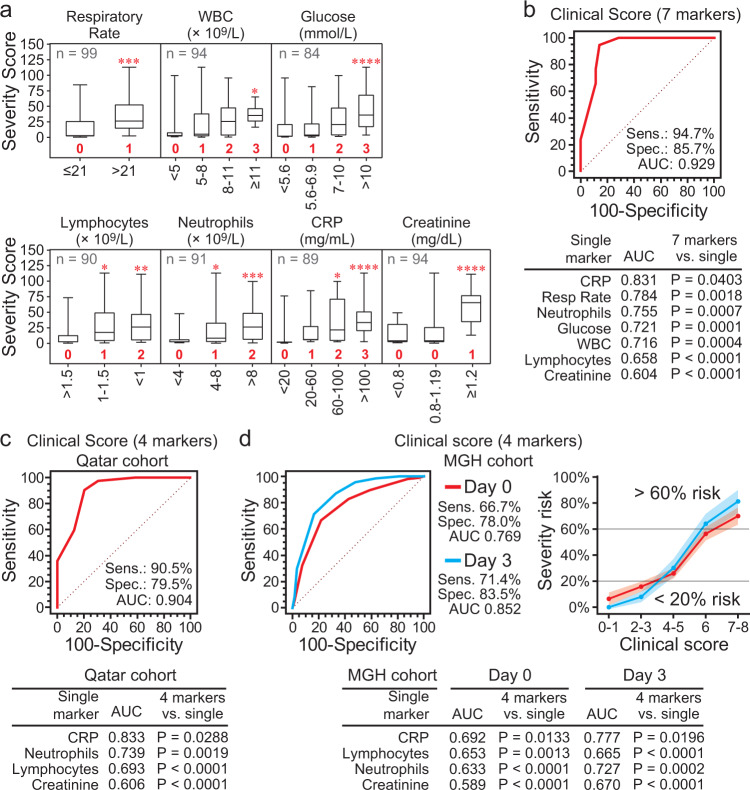
COVID-19 complications still present a huge burden on healthcare systems and warrant predictive risk models to triage patients and inform early intervention. Here, we profile 893 plasma proteins from 50 severe and 50 mild-moderate COVID-19 patients, and 50 healthy controls, and show that 375 proteins are differentially expressed in the plasma of severe COVID-19 patients. These differentially expressed plasma proteins are implicated in the pathogenesis of COVID-19 and present targets for candidate drugs to prevent or treat severe complications. Based on the plasma proteomics and clinical lab tests, we also report a 12-plasma protein signature and a model of seven routine clinical tests that validate in an independent cohort as early risk predictors of COVID-19 severity and patient survival. The risk predictors and candidate drugs described in our study can be used and developed for personalized management of SARS-CoV-2 infected patients.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- ISARIC Clinical Characterisation Group, et al. ISARIC Clinical Data Report issued: 14 July 2021. medRxiv, 2020.2007.2017.20155218. Preprint at 10.1101/2020.07.17.20155218 (2021).
-
- Uhlen M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
