

Supergene origin and maintenance in Atlantic cod
- PMID: 35177802
- PMCID: PMC8986531
- DOI: 10.1038/s41559-022-01661-x
Supergene origin and maintenance in Atlantic cod
Abstract
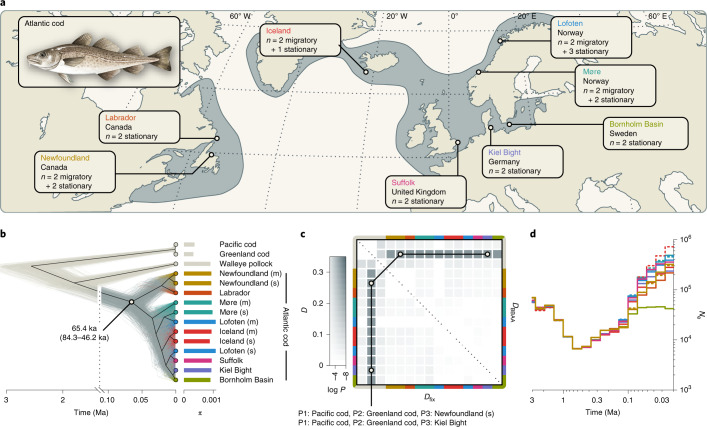
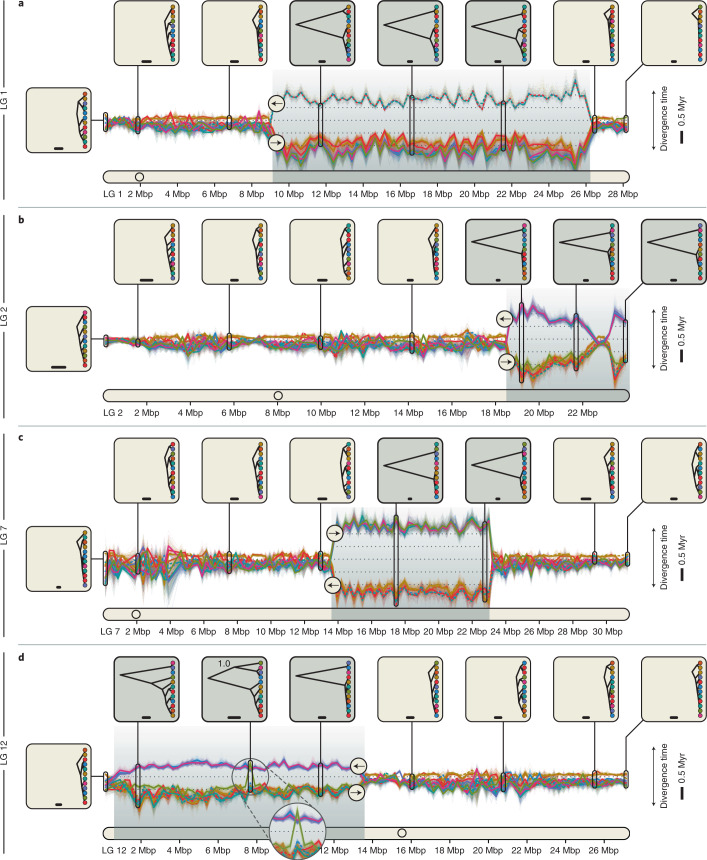
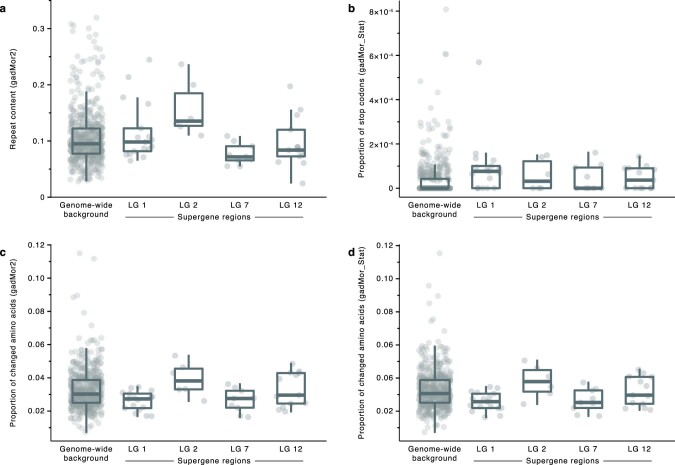
Supergenes are sets of genes that are inherited as a single marker and encode complex phenotypes through their joint action. They are identified in an increasing number of organisms, yet their origins and evolution remain enigmatic. In Atlantic cod, four megabase-scale supergenes have been identified and linked to migratory lifestyle and environmental adaptations. Here we investigate the origin and maintenance of these four supergenes through analysis of whole-genome-sequencing data, including a new long-read-based genome assembly for a non-migratory Atlantic cod individual. We corroborate the finding that chromosomal inversions underlie all four supergenes, and we show that they originated at different times between 0.40 and 1.66 million years ago. We reveal gene flux between supergene haplotypes where migratory and stationary Atlantic cod co-occur and conclude that this gene flux is driven by gene conversion, on the basis of an increase in GC content in exchanged sites. Additionally, we find evidence for double crossover between supergene haplotypes, leading to the exchange of an ~275 kilobase fragment with genes potentially involved in adaptation to low salinity in the Baltic Sea. Our results suggest that supergenes can be maintained over long timescales in the same way as hybridizing species, through the selective purging of introduced genetic variation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





Comment in
-
Evolution of cod supergenes.Nat Ecol Evol. 2022 Apr;6(4):355-356. doi: 10.1038/s41559-022-01662-w. Nat Ecol Evol. 2022. PMID: 35177801 No abstract available.
References
-
- Yan Z, et al. Evolution of a supergene that regulates a trans-species social polymorphism. Nat. Ecol. Evol. 2020;4:210–249. - PubMed
-
- Lamichhaney S, et al. Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax) Nat. Genet. 2016;48:84–88. - PubMed
-
- Li J, et al. Genetic architecture and evolution of the S locus supergene in Primula vulgaris. Nat. Plants. 2016;2:16188. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
