

Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank
- PMID: 35177841
- PMCID: PMC8930703
- DOI: 10.1038/s41588-021-01011-w
Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank
Abstract
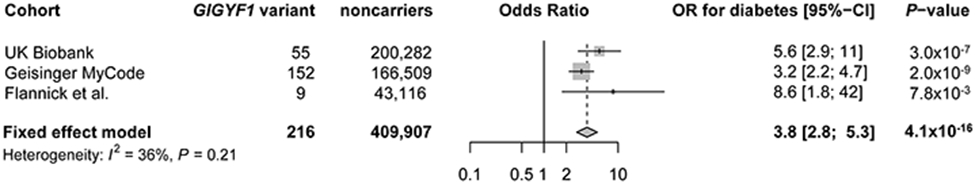
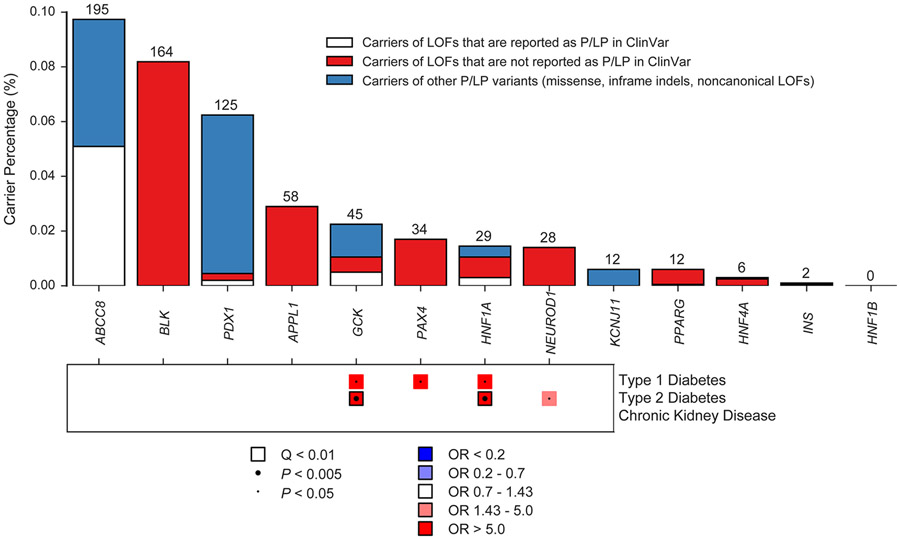
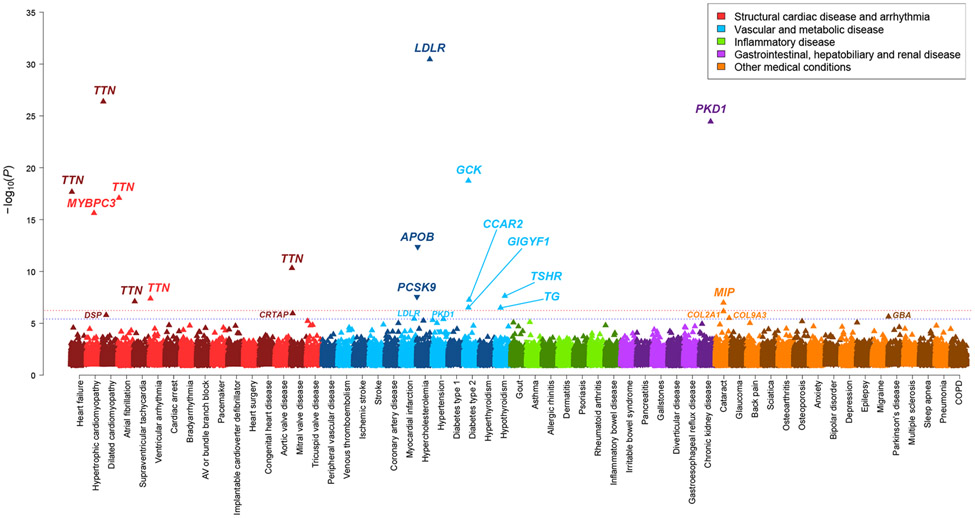
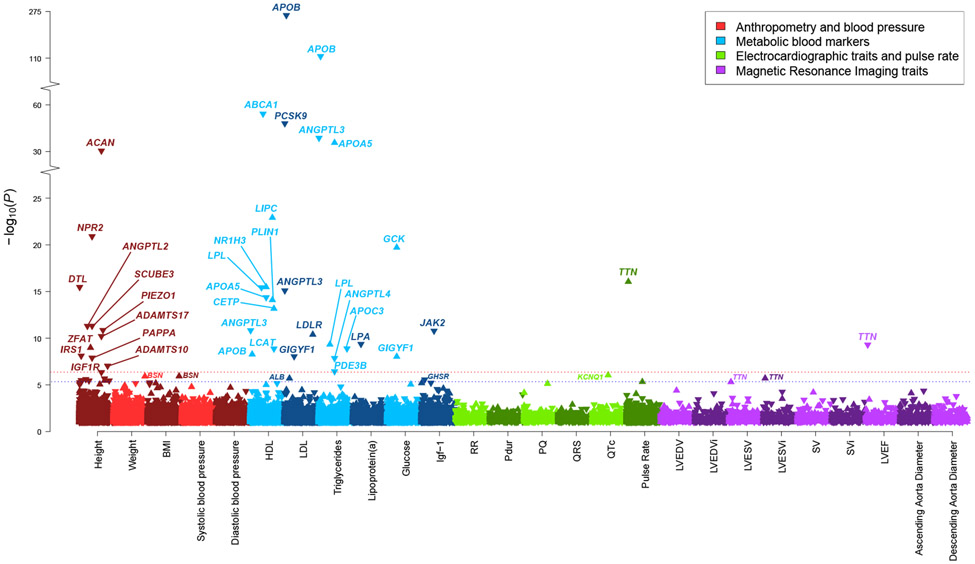
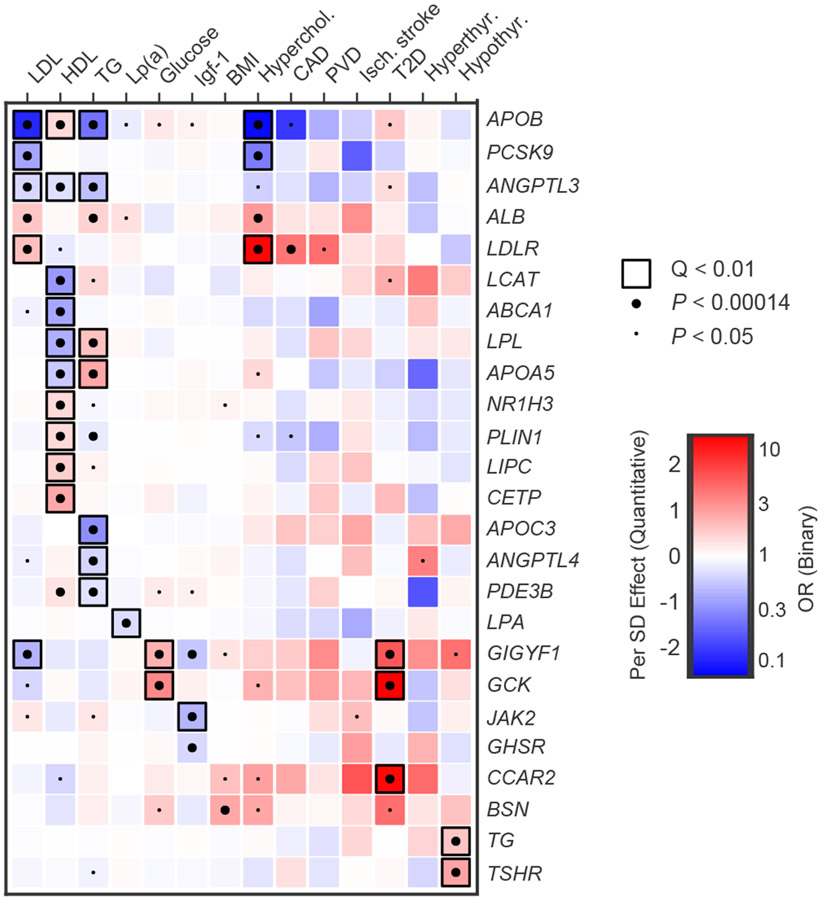
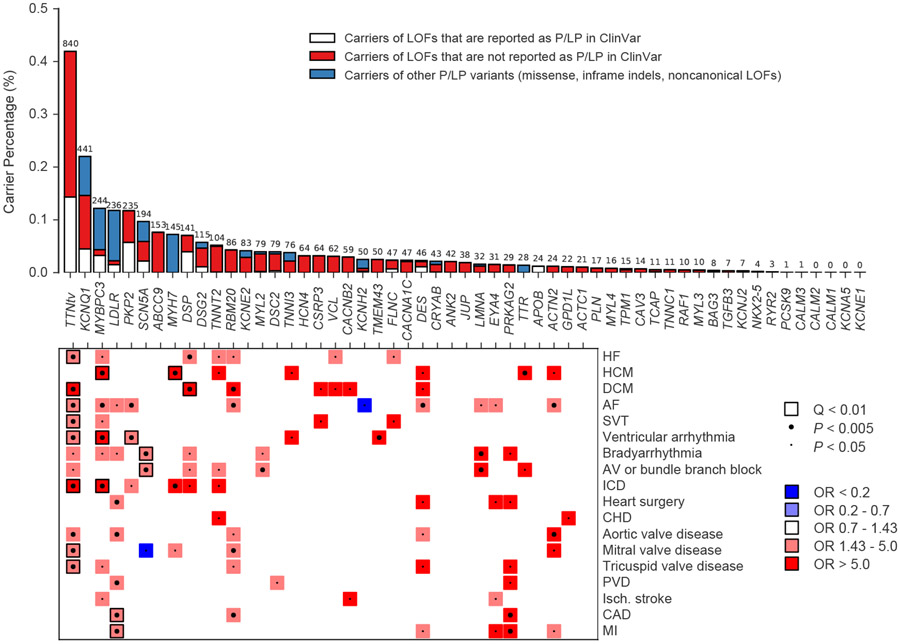
Cardiometabolic diseases are the leading cause of death worldwide. Despite a known genetic component, our understanding of these diseases remains incomplete. Here, we analyzed the contribution of rare variants to 57 diseases and 26 cardiometabolic traits, using data from 200,337 UK Biobank participants with whole-exome sequencing. We identified 57 gene-based associations, with broad replication of novel signals in Geisinger MyCode. There was a striking risk associated with mutations in known Mendelian disease genes, including MYBPC3, LDLR, GCK, PKD1 and TTN. Many genes showed independent convergence of rare and common variant evidence, including an association between GIGYF1 and type 2 diabetes. We identified several large effect associations for height and 18 unique genes associated with blood lipid or glucose levels. Finally, we found that between 1.0% and 2.4% of participants carried rare potentially pathogenic variants for cardiometabolic disorders. These findings may facilitate studies aimed at therapeutics and screening of these common disorders.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
Methods-only References
-
- Heinze G A comparative investigation of methods for logistic regression with separated or nearly separated data. Statistics in Medicine 25, 4216–4226 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
