

Periodontal Infection Aggravates C1q-Mediated Microglial Activation and Synapse Pruning in Alzheimer's Mice
- PMID: 35178049
- PMCID: PMC8845011
- DOI: 10.3389/fimmu.2022.816640
Periodontal Infection Aggravates C1q-Mediated Microglial Activation and Synapse Pruning in Alzheimer's Mice
Abstract
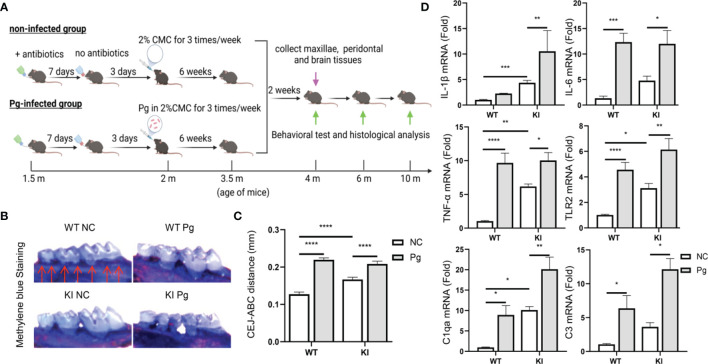
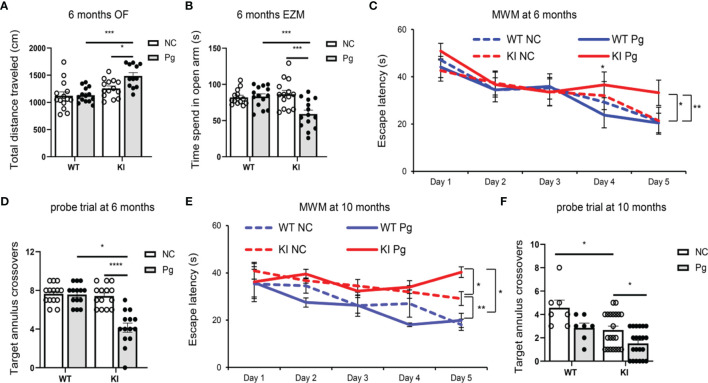
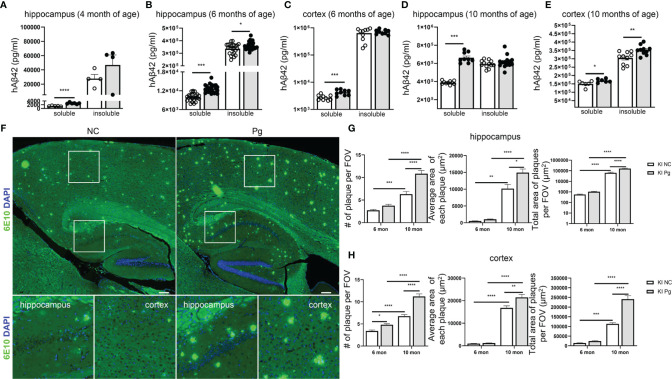
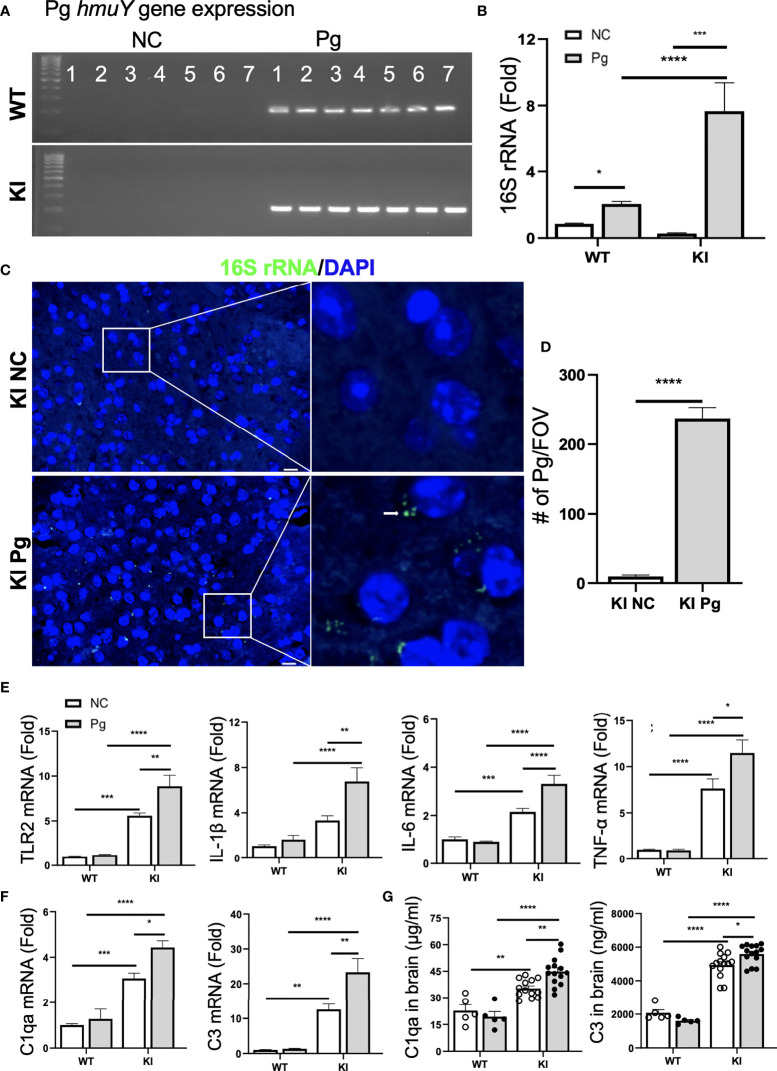
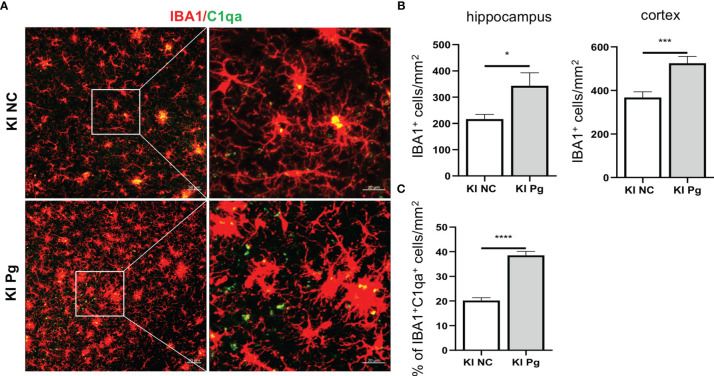
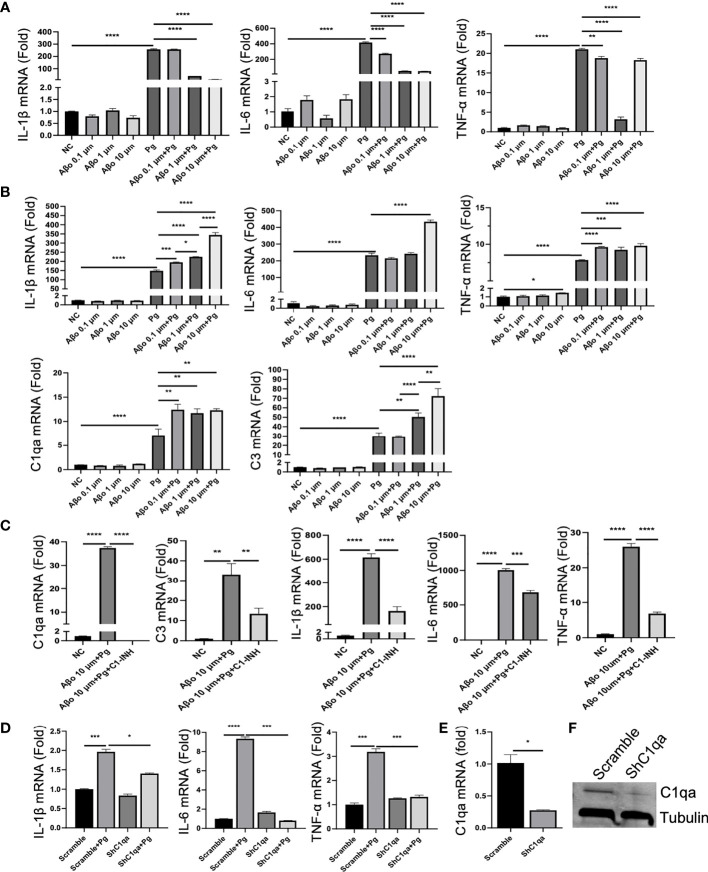
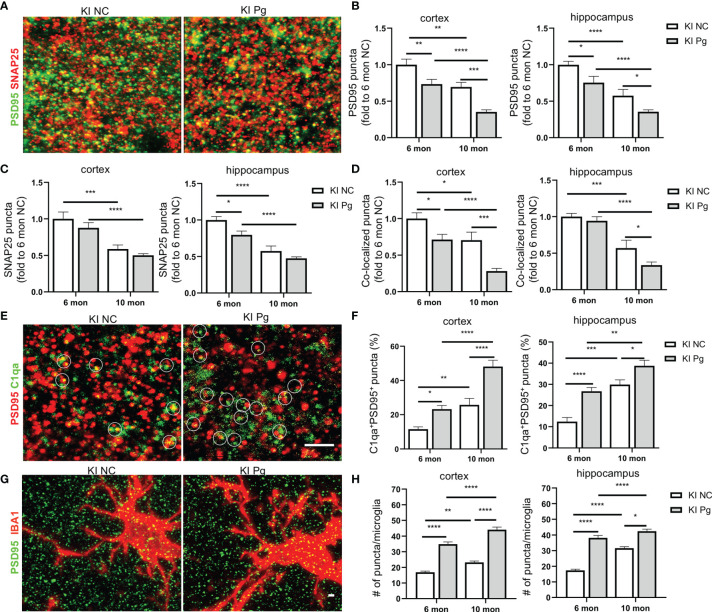
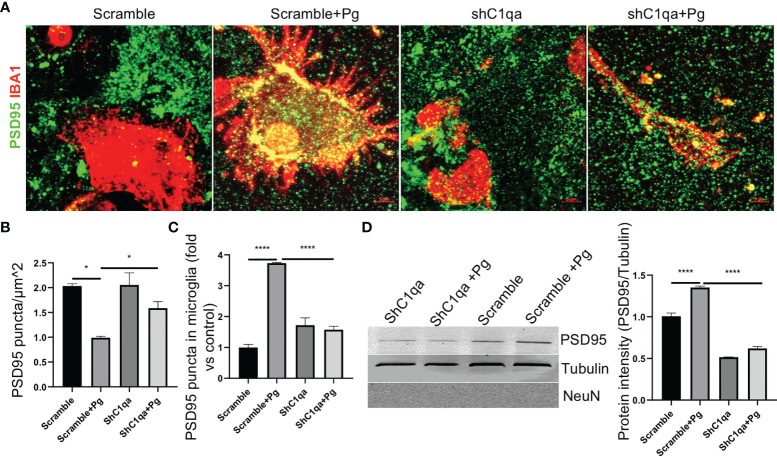
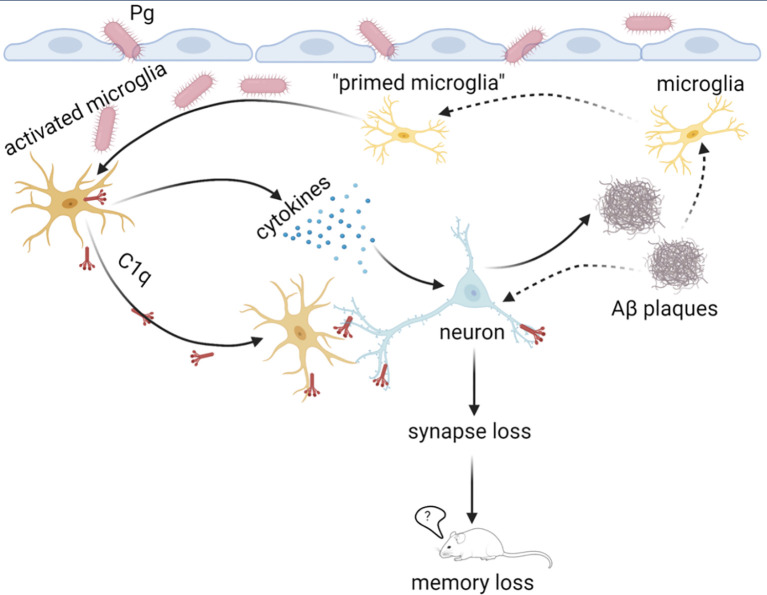
Periodontitis is a dysbiotic infectious disease that leads to the destruction of tooth supporting tissues. There is increasing evidence that periodontitis may affect the development and severity of Alzheimer's disease (AD). However, the mechanism(s) by which periodontal infection impacts the neurodegenerative process in AD remains unclear. In the present study, using an amyloid precursor protein (APP) knock-in (App KI) AD mouse model, we showed that oral infection with Porphyromonas gingivalis (Pg), a keystone pathogen of periodontitis, worsened behavioral and cognitive impairment and accelerated amyloid beta (Aβ) accumulation in AD mice, thus unquestionably and significantly aggravating AD. We also provide new evidence that the neuroinflammatory status established by AD, is greatly complicated by periodontal infection and the consequential entry of Pg into the brain via Aβ-primed microglial activation, and that Pg-induced brain overactivation of complement C1q is critical for periodontitis-associated acceleration of AD progression by amplifying microglial activation, neuroinflammation, and tagging synapses for microglial engulfment. Our study renders support for the importance of periodontal infection in the innate immune regulation of AD and the possibility of targeting microbial etiology and periodontal treatment to ameliorate the clinical manifestation of AD and lower AD prevalence.
Keywords: Alzheimer’s disease; Periodontitis; Porphyromonas gingivalis; complement C1q; microglia; synapse loss.
Copyright © 2022 Hao, Li, Li, Katz, Michalek, Barnum, Pozzo-Miller, Saito, Saido, Wang, Roberson and Zhang.
Conflict of interest statement
SB is the CSO & Co-Founder of CNine Biosolutions, LLC. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice.PLoS One. 2018 Oct 3;13(10):e0204941. doi: 10.1371/journal.pone.0204941. eCollection 2018. PLoS One. 2018. PMID: 30281647 Free PMC article.
-
Intestinal homeostasis disrupted by Periodontitis exacerbates Alzheimer's Disease in APP/PS1 mice.J Neuroinflammation. 2024 Oct 18;21(1):263. doi: 10.1186/s12974-024-03256-8. J Neuroinflammation. 2024. PMID: 39425119 Free PMC article.
-
GSK3β is involved in promoting Alzheimer's disease pathologies following chronic systemic exposure to Porphyromonas gingivalis lipopolysaccharide in amyloid precursor proteinNL-F/NL-F knock-in mice.Brain Behav Immun. 2021 Nov;98:1-12. doi: 10.1016/j.bbi.2021.08.213. Epub 2021 Aug 13. Brain Behav Immun. 2021. PMID: 34391814 Free PMC article.
-
Microglial Cathepsin B and Porphyromonas gingivalis Gingipains as Potential Therapeutic Targets for Sporadic Alzheimer's Disease.CNS Neurol Disord Drug Targets. 2020;19(7):495-502. doi: 10.2174/1871527319666200708125130. CNS Neurol Disord Drug Targets. 2020. PMID: 32640970 Review.
-
Effects of CX3CR1 and Fractalkine Chemokines in Amyloid Beta Clearance and p-Tau Accumulation in Alzheimer's Disease (AD) Rodent Models: Is Fractalkine a Systemic Biomarker for AD?Curr Alzheimer Res. 2016;13(4):403-12. doi: 10.2174/1567205013666151116125714. Curr Alzheimer Res. 2016. PMID: 26567742 Review.
Cited by
-
Association between periodontitis and Alzheimer's disease: A narrative review.IBRO Neurosci Rep. 2025 Jan 17;18:360-365. doi: 10.1016/j.ibneur.2024.12.004. eCollection 2025 Jun. IBRO Neurosci Rep. 2025. PMID: 40060035 Free PMC article. Review.
-
Alzheimer's Disease and Porphyromonas gingivalis: Exploring the Links.Life (Basel). 2025 Jan 14;15(1):96. doi: 10.3390/life15010096. Life (Basel). 2025. PMID: 39860036 Free PMC article. Review.
-
Exploring the Link Between Periodontitis and Alzheimer's Disease-Could a Nanoparticulate Vaccine Break It?Pharmaceutics. 2025 Jan 21;17(2):141. doi: 10.3390/pharmaceutics17020141. Pharmaceutics. 2025. PMID: 40006510 Free PMC article. Review.
-
Oral polymicrobial communities: Assembly, function, and impact on diseases.Cell Host Microbe. 2023 Apr 12;31(4):528-538. doi: 10.1016/j.chom.2023.02.009. Epub 2023 Mar 17. Cell Host Microbe. 2023. PMID: 36933557 Free PMC article. Review.
-
Oral administration of Porphyromonas gingivalis to mice with diet-induced obesity impairs cognitive function associated with microglial activation in the brain.J Oral Microbiol. 2024 Nov 14;16(1):2419155. doi: 10.1080/20002297.2024.2419155. eCollection 2024. J Oral Microbiol. 2024. PMID: 39553478 Free PMC article.
References
-
- Association . As. 2020 Alzheimer’s Disease Facts and Figures. Alzheimers Dement (2020) 16:391–460. doi: 10.1002/alz.12068 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases