

Non-cell-autonomous disruption of nuclear architecture as a potential cause of COVID-19-induced anosmia
- PMID: 35180380
- PMCID: PMC8808699
- DOI: 10.1016/j.cell.2022.01.024
Non-cell-autonomous disruption of nuclear architecture as a potential cause of COVID-19-induced anosmia
Abstract
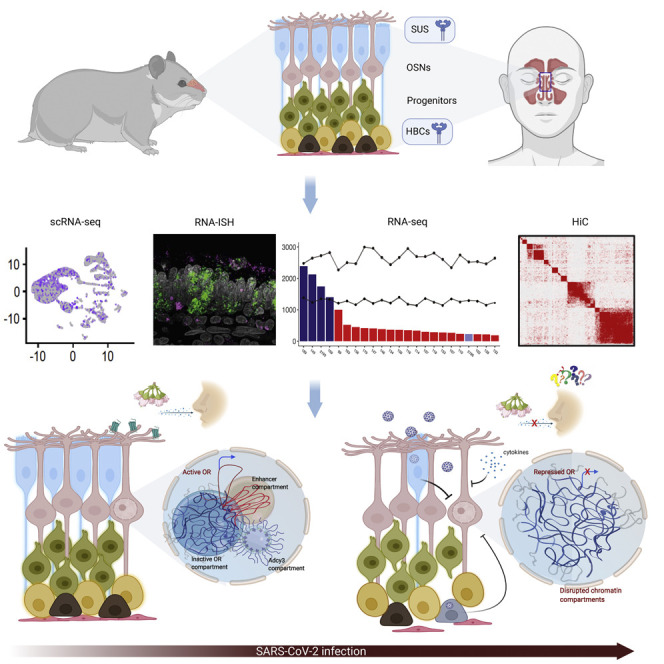
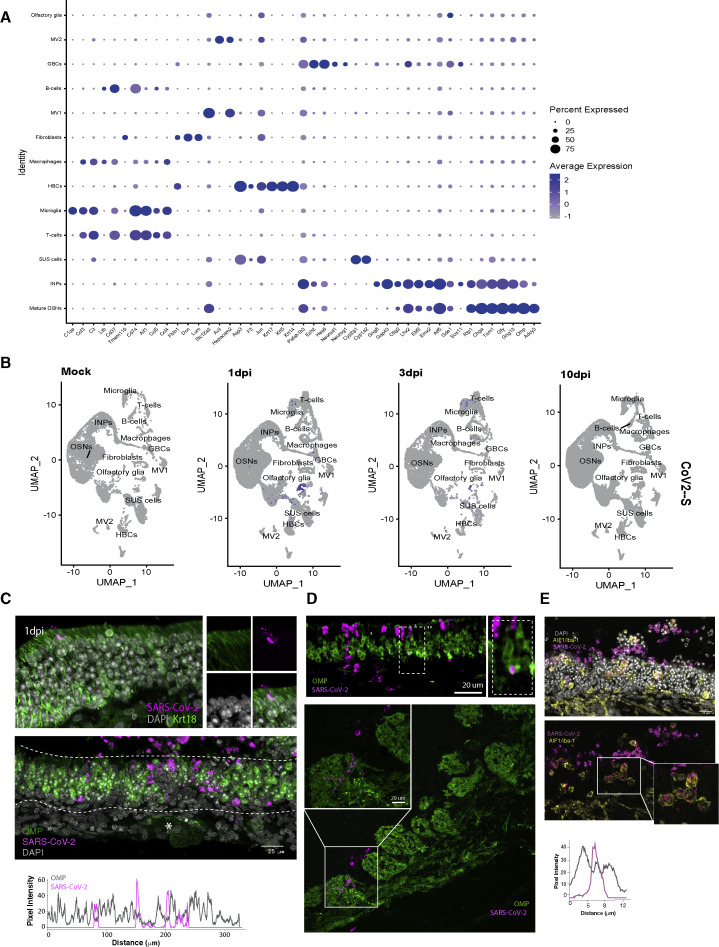
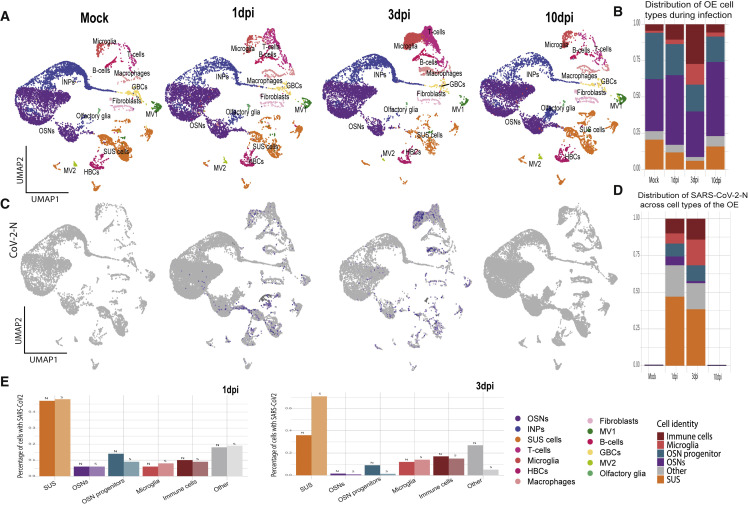
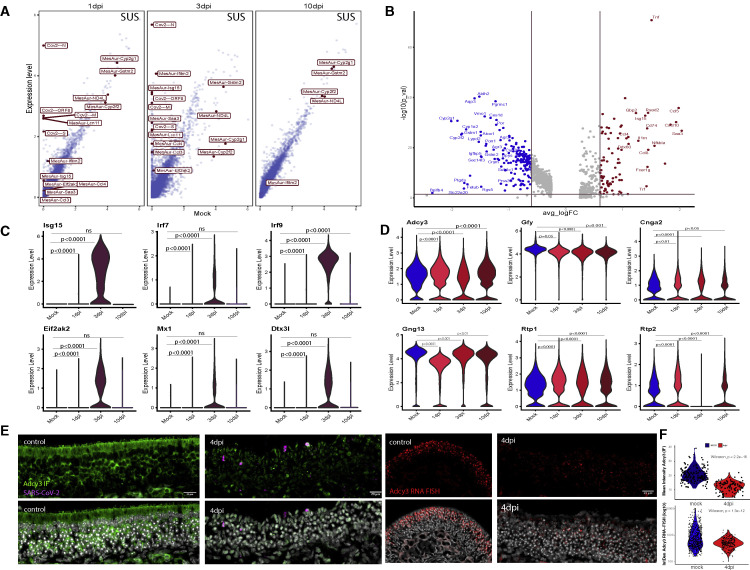
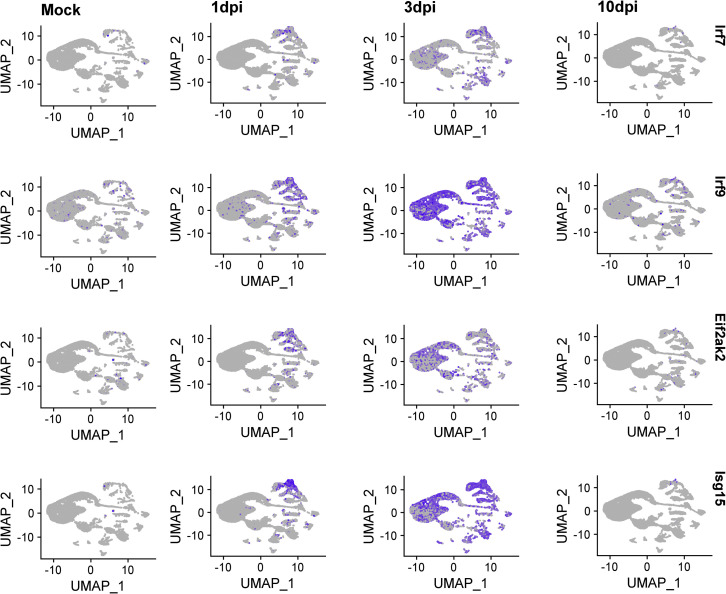
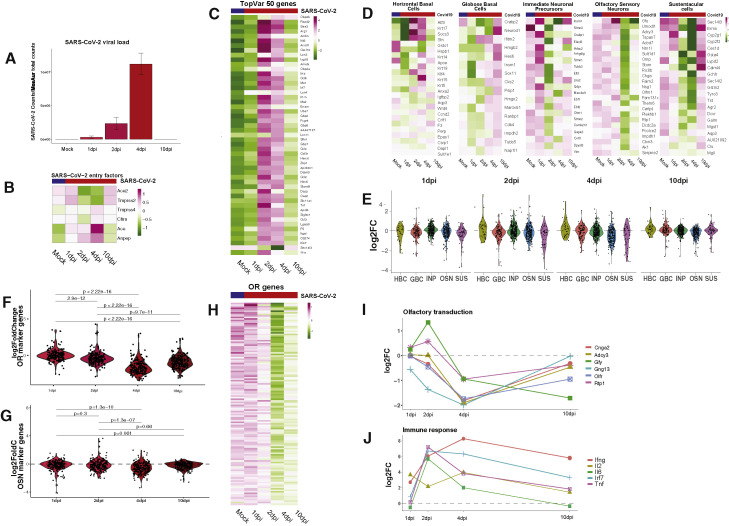
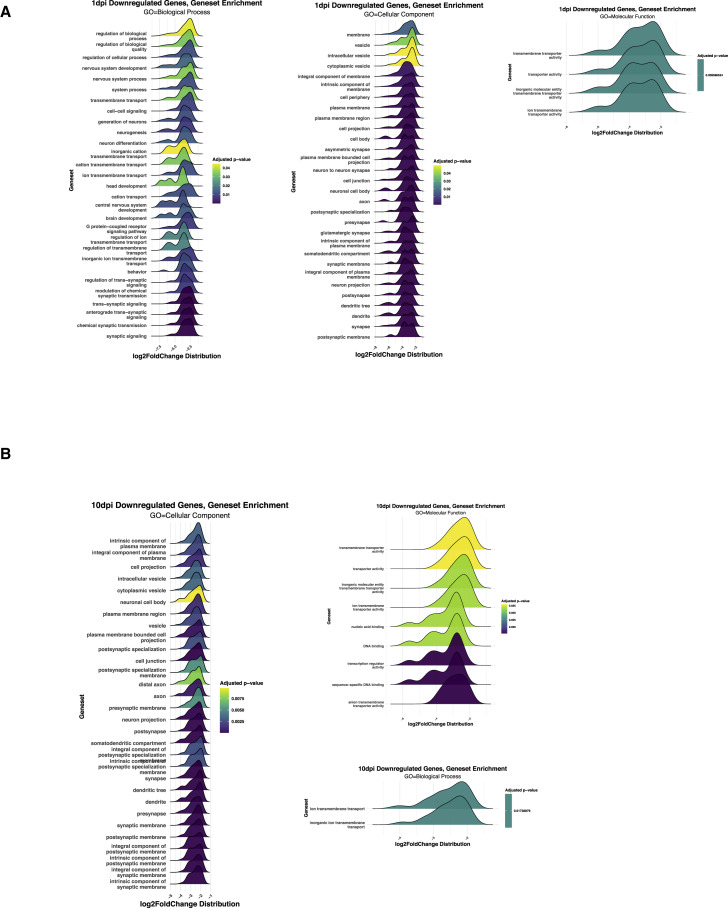
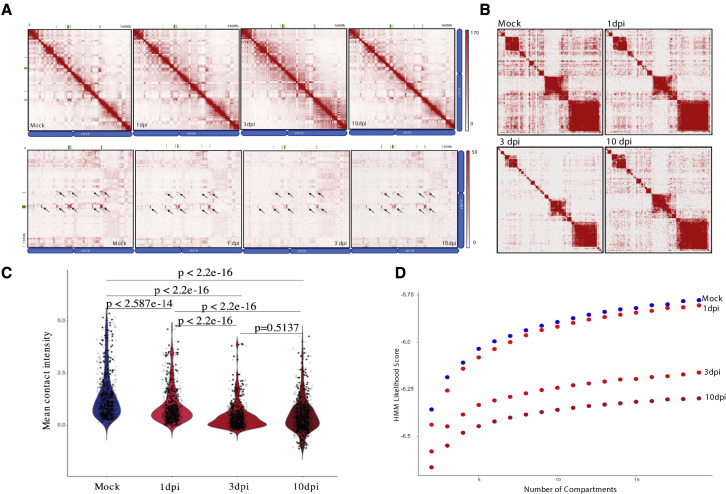
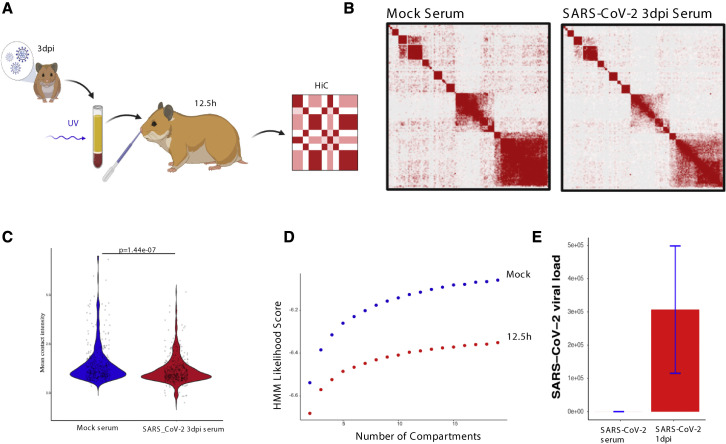
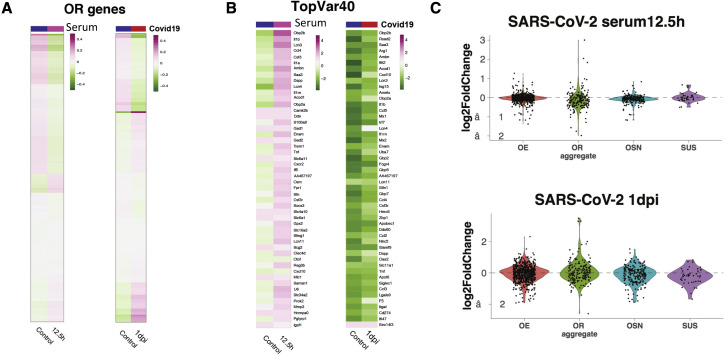
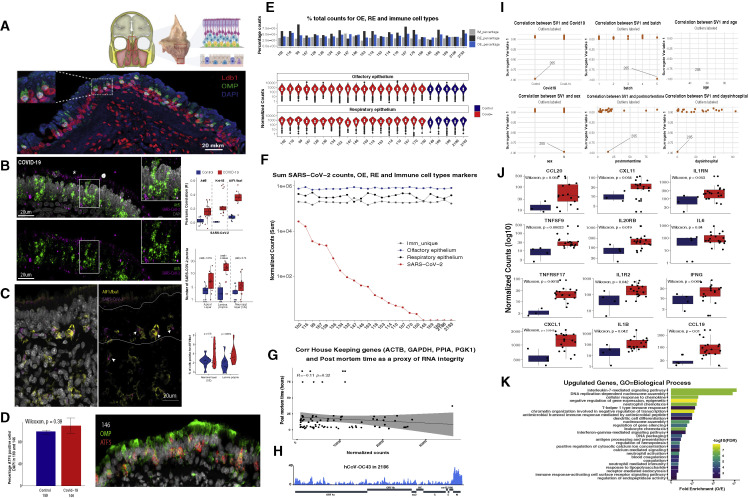
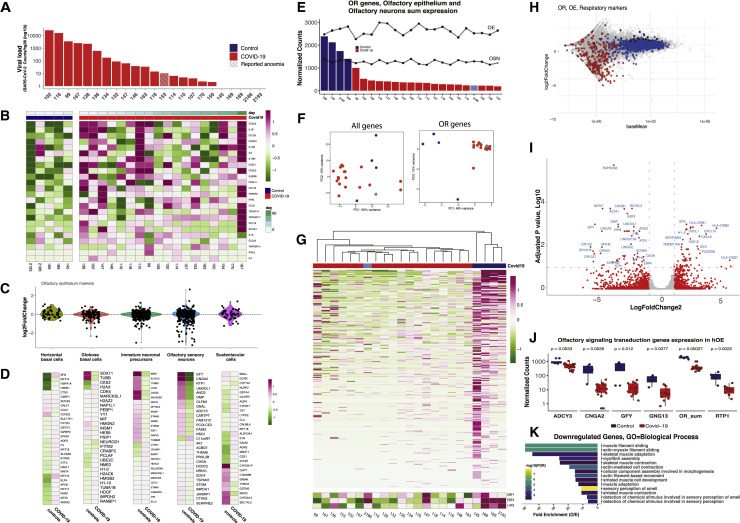
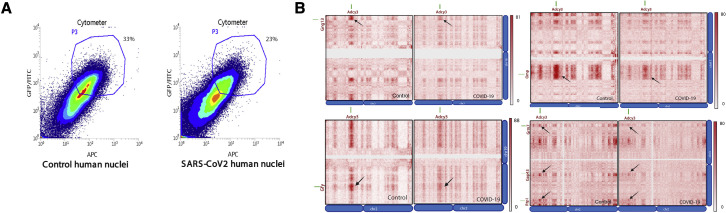
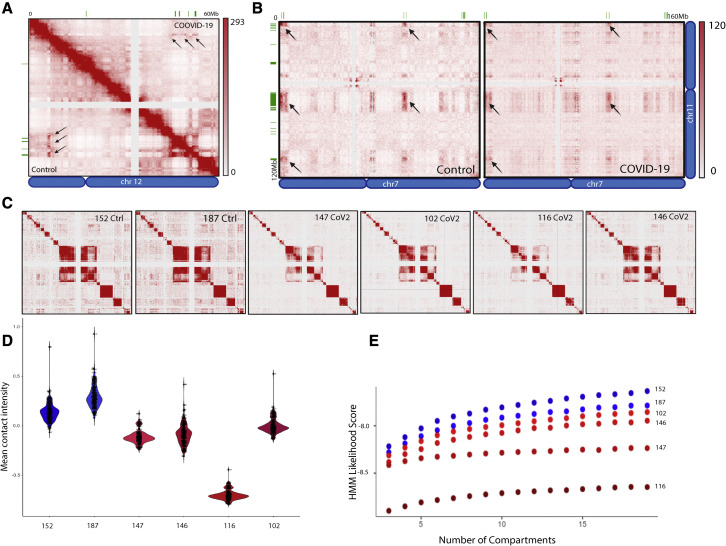
SARS-CoV-2 infects less than 1% of cells in the human body, yet it can cause severe damage in a variety of organs. Thus, deciphering the non-cell-autonomous effects of SARS-CoV-2 infection is imperative for understanding the cellular and molecular disruption it elicits. Neurological and cognitive defects are among the least understood symptoms of COVID-19 patients, with olfactory dysfunction being their most common sensory deficit. Here, we show that both in humans and hamsters, SARS-CoV-2 infection causes widespread downregulation of olfactory receptors (ORs) and of their signaling components. This non-cell-autonomous effect is preceded by a dramatic reorganization of the neuronal nuclear architecture, which results in dissipation of genomic compartments harboring OR genes. Our data provide a potential mechanism by which SARS-CoV-2 infection alters the cellular morphology and the transcriptome of cells it cannot infect, offering insight to its systemic effects in olfaction and beyond.
Keywords: COVID-19; anosmia; nuclear architecture.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Update of
-
Disruption of nuclear architecture as a cause of COVID-19 induced anosmia.bioRxiv [Preprint]. 2021 Feb 9:2021.02.09.430314. doi: 10.1101/2021.02.09.430314. bioRxiv. 2021. Update in: Cell. 2022 Mar 17;185(6):1052-1064.e12. doi: 10.1016/j.cell.2022.01.024. PMID: 33594368 Free PMC article. Updated. Preprint.
Comment in
-
SARS-CoV-2 sensory loss.Nat Rev Microbiol. 2022 Apr;20(4):190. doi: 10.1038/s41579-022-00707-y. Nat Rev Microbiol. 2022. PMID: 35177830 Free PMC article.
-
Can't smell the virus: SARS-CoV-2, chromatin organization, and anosmia.Dev Cell. 2022 May 9;57(9):1081-1082. doi: 10.1016/j.devcel.2022.04.015. Dev Cell. 2022. PMID: 35537401 Free PMC article.
References
-
- Albers M.W., Tabert M.H., Devanand D.P. Olfactory dysfunction as a predictor of neurodegenerative disease. Curr. Neurol. Neurosci. Rep. 2006;6:379–386. - PubMed
-
- Baharlou H., Canete N.P., Bertram K.M., Sandgren K.J., Cunningham A.L., Harman A.N., Patrick E. AFid: A tool for automated identification and exclusion of autofluorescent objects from microscopy images. bioRxiv. 2019 - PubMed
-
- Bashkirova E., Monahan K., Campbell C.E., Osinski J.M., Tan L., Schieren I., Barnea G., Xie X.S., Gronostajski R.M., Lomvardas S., et al. Homeotic regulation of olfactory receptor choice via NFI-dependent heterochromatic silencing and genomic compartmentalization. bioRxiv. 2020
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
