

Systemic Chemotherapies Retain Antitumor Activity in Desmoid Tumors Independent of Specific Mutations in CTNNB1 or APC: A Multi-institutional Retrospective Study
- PMID: 35180772
- PMCID: PMC9475245
- DOI: 10.1158/1078-0432.CCR-21-4504
Systemic Chemotherapies Retain Antitumor Activity in Desmoid Tumors Independent of Specific Mutations in CTNNB1 or APC: A Multi-institutional Retrospective Study
Abstract
Purpose: Determine whether specific CTNNB1 or APC mutations in patients with desmoid tumor were associated with differences in clinical responses to systemic treatments.
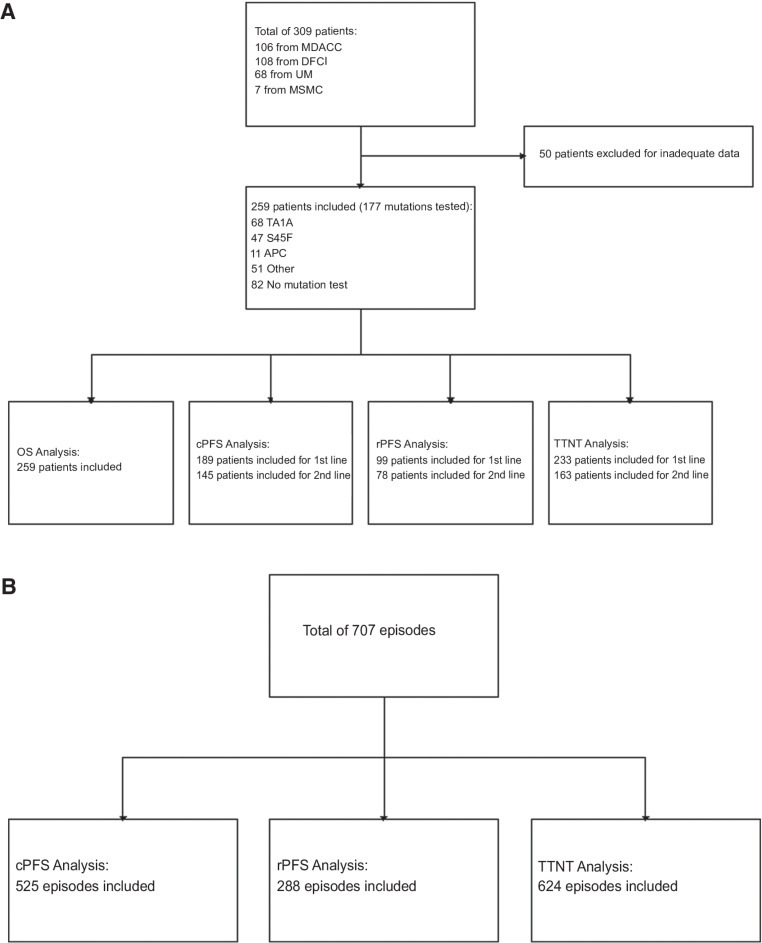
Experimental design: We established a multi-institutional dataset of previously treated patients with desmoid tumor across four U.S. sarcoma centers, including demographic and clinicopathologic characteristics, treatment regimens, and clinical and radiographic responses. CTNNB1 or APC mutation status was determined from prior pathology records, or archival tissue was requested and analyzed by Sanger sequencing and/or next-generation sequencing. Evaluable patients with mutation results were analyzed to determine clinical progression-free survival (cPFS), RECIST 1.1 PFS (rPFS), time to next treatment (TTNT), and overall survival (OS). Kaplan-Meier analysis and Cox proportional hazards regression were performed to identify differences in cPFS, rPFS, TTNT, and OS by mutation subtype, desmoid tumor location, and treatment regimen.
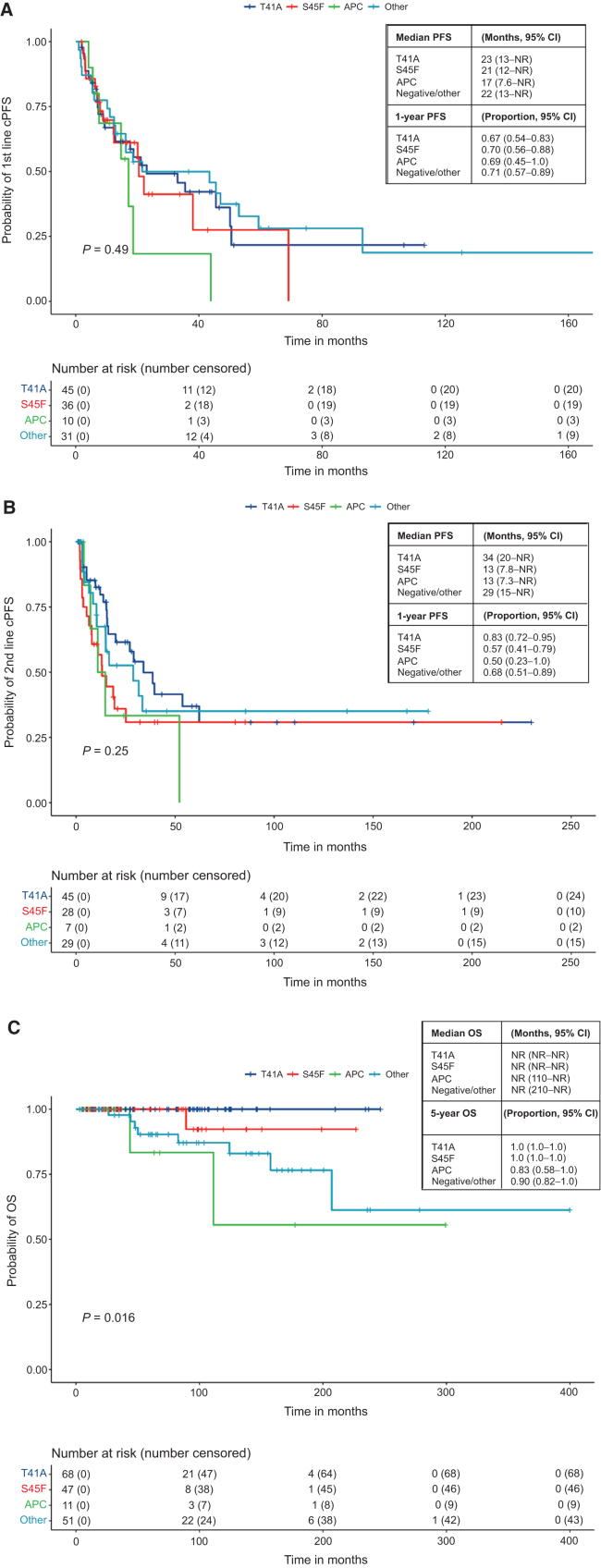
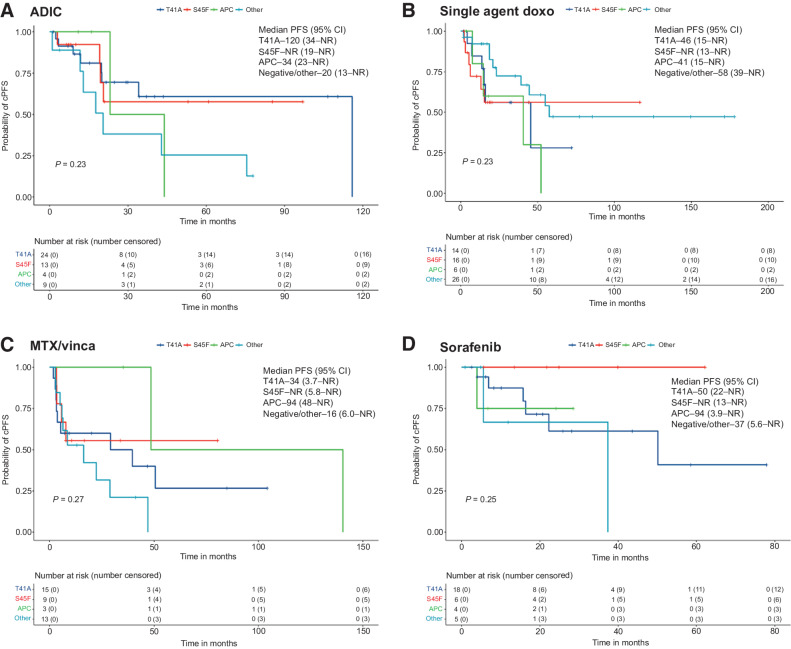
Results: A total of 259 evaluable patients were analyzed for at least one of the survival outcomes, with 177 patients having mutation data. First- and second-line cPFS, rPFS, and TTNT were not significantly affected by mutation subtype; however, APC-mutant desmoid tumors demonstrated nonstatistically significant inferior outcomes. Extremity/trunk desmoid tumor location and treatment with doxorubicin-based, methotrexate/vinca alkaloids and sorafenib regimens were associated with better clinical outcomes compared with surgery or "other" therapies, including estrogen-receptor blockade and imatinib. OS was significantly worse with APC or CTNNB1 negative/other mutations.
Conclusions: Mutation subtype did not affect responses to specific systemic therapies. APC mutations and nonextremity desmoid tumor locations remain prognostic for worse outcomes, and earlier initiation of systemic therapy for these higher-risk desmoid tumors should be prospectively evaluated. See related commentary by Greene and Van Tine, p. 3911.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
-
Are the Pieces Starting to Come Together for Management of Desmoid Tumors?Clin Cancer Res. 2022 Sep 15;28(18):3911-3913. doi: 10.1158/1078-0432.CCR-22-0620. Clin Cancer Res. 2022. PMID: 35819317
References
-
- WHO classification of tumours of soft tissue and bone. 4th ed.Lyon, France: WHO, International Agency for Research on Cancer IARC; 2013.
-
- Reitamo JJ, Häyry P, Nykyri E, Saxen E. The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the finnish population. Am J Clin Pathol 1982;77:665–73. - PubMed
-
- Reitamo JJ, Scheinin TM, Hayry P. The desmoid syndrome. New aspects in the cause, pathogenesis and treatment of the desmoid tumor. Am J Surg 1986;151:230–7. - PubMed
-
- Nieuwenhuis MH, Casparie M, Mathus-Vliegen LMH, Dekkers OM, Hogendoorn PCW, Vasen HFA. A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer 2011;129:256–61. - PubMed
-
- Salas S, Dufresne A, Bui B, Blay J-Y, Terrier P, Ranchere-Vince D, et al. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: a wait-and-see policy according to tumor presentation. J Clin Oncol 2011;29:3553–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
