

GeneTerpret: a customizable multilayer approach to genomic variant prioritization and interpretation
- PMID: 35180879
- PMCID: PMC8857790
- DOI: 10.1186/s12920-022-01166-3
GeneTerpret: a customizable multilayer approach to genomic variant prioritization and interpretation
Abstract
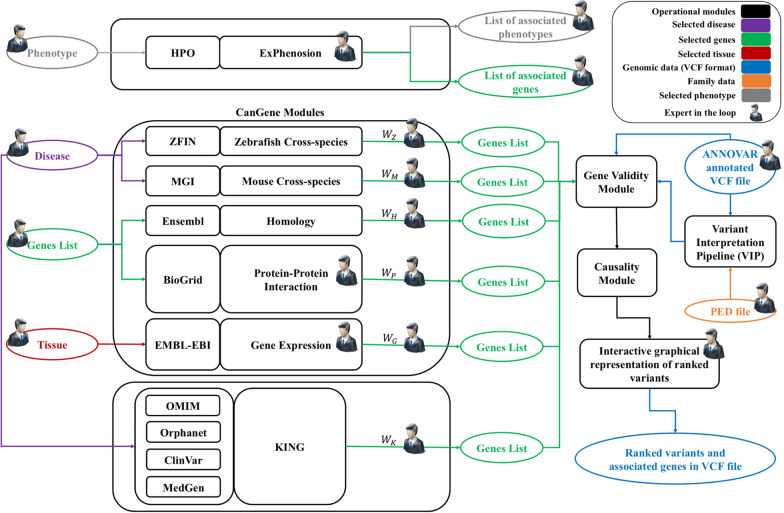
Background: Variant interpretation is the main bottleneck in medical genomic sequencing efforts. This usually involves genome analysts manually searching through a multitude of independent databases, often with the aid of several, mostly independent, computational tools. To streamline variant interpretation, we developed the GeneTerpret platform which collates data from current interpretation tools and databases, and applies a phenotype-driven query to categorize the variants identified in the genome(s). The platform assigns quantitative validity scores to genes by query and assembly of the genotype-phenotype data, sequence homology, molecular interactions, expression data, and animal models. It also uses the American College of Medical Genetics and Genomics (ACMG) criteria to categorize variants into five tiers of pathogenicity. The final output is a prioritized list of potentially causal variants/genes.
Results: We tested GeneTerpret by comparing its performance to expert-curated genes (ClinGen's gene-validity database) and variant pathogenicity reports (DECIPHER database). Output from GeneTerpret was 97.2% and 83.5% concordant with the expert-curated sources, respectively. Additionally, similar concordance was observed when GeneTerpret's performance was compared with our internal expert-interpreted clinical datasets.
Conclusions: GeneTerpret is a flexible platform designed to streamline the genome interpretation process, through a unique interface, with improved ease, speed and accuracy. This modular and customizable system allows the user to tailor the component-programs in the analysis process to their preference. GeneTerpret is available online at https://geneterpret.com .
Keywords: Bioinformatic application; Causative variants; Disease gene validity; Gene prioritization; Genome interpretation; Genomic variants; Genotype–phenotype correlation; Variant pathogenicity.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
