

PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease
- PMID: 35184140
- PMCID: PMC8858315
- DOI: 10.1038/s41419-022-04613-2
PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease
Abstract
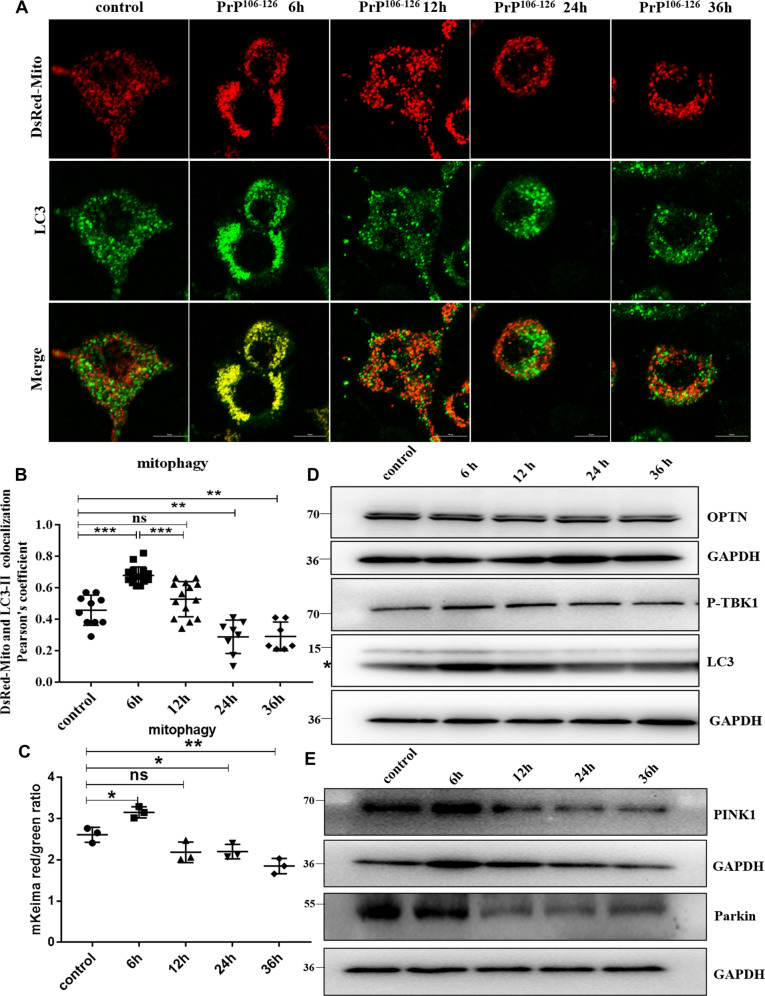
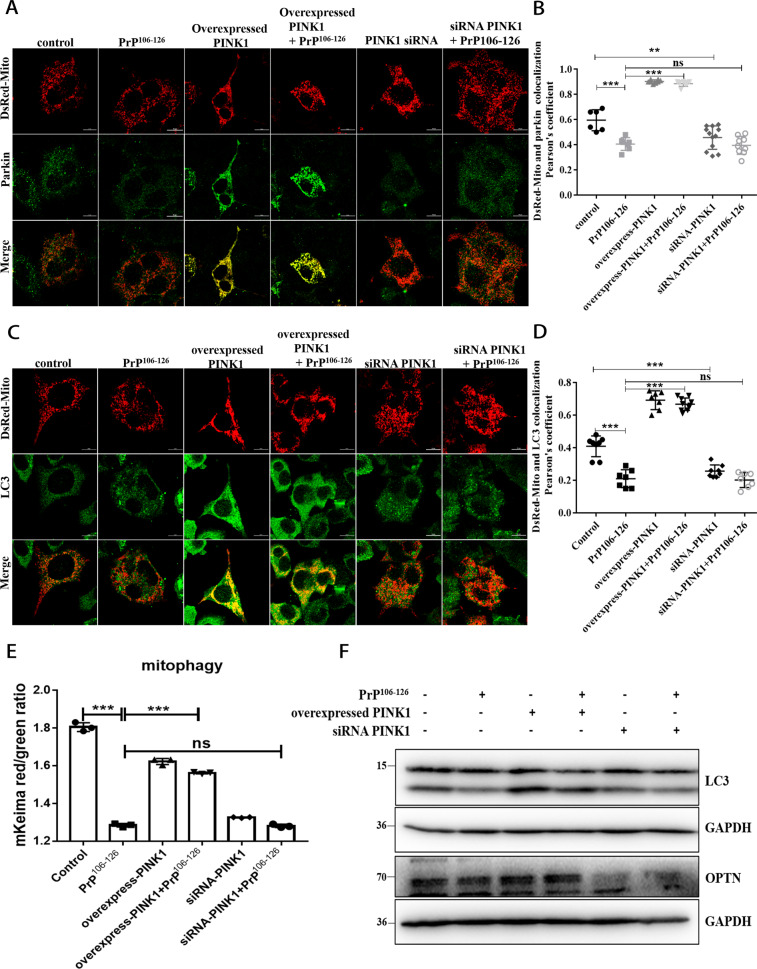
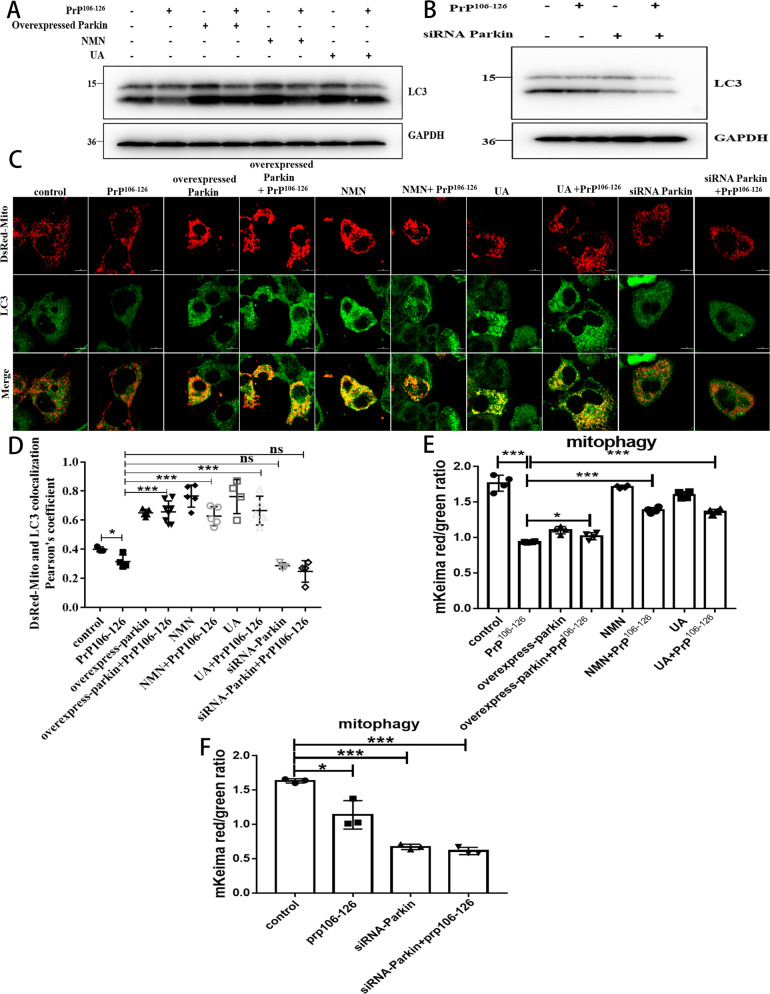
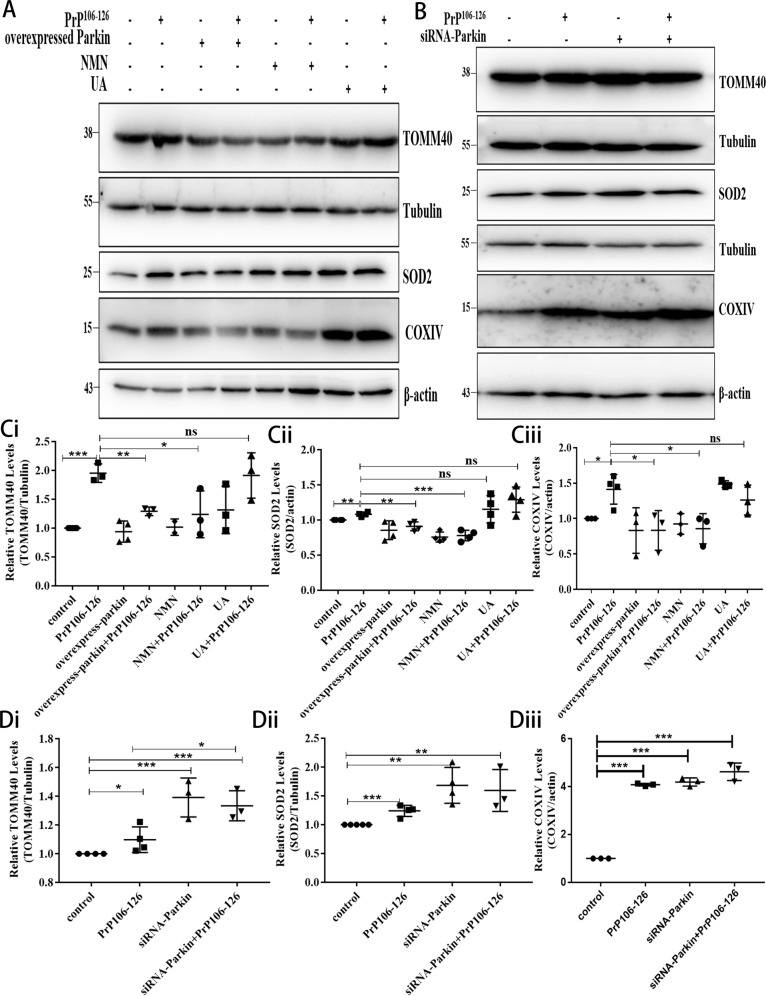
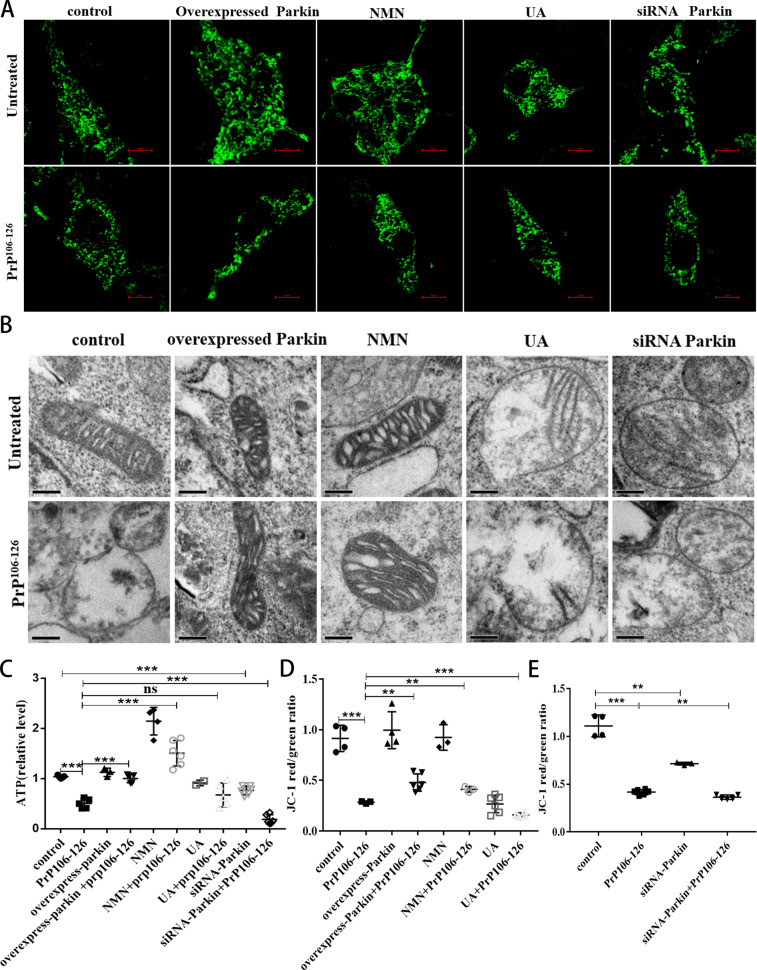
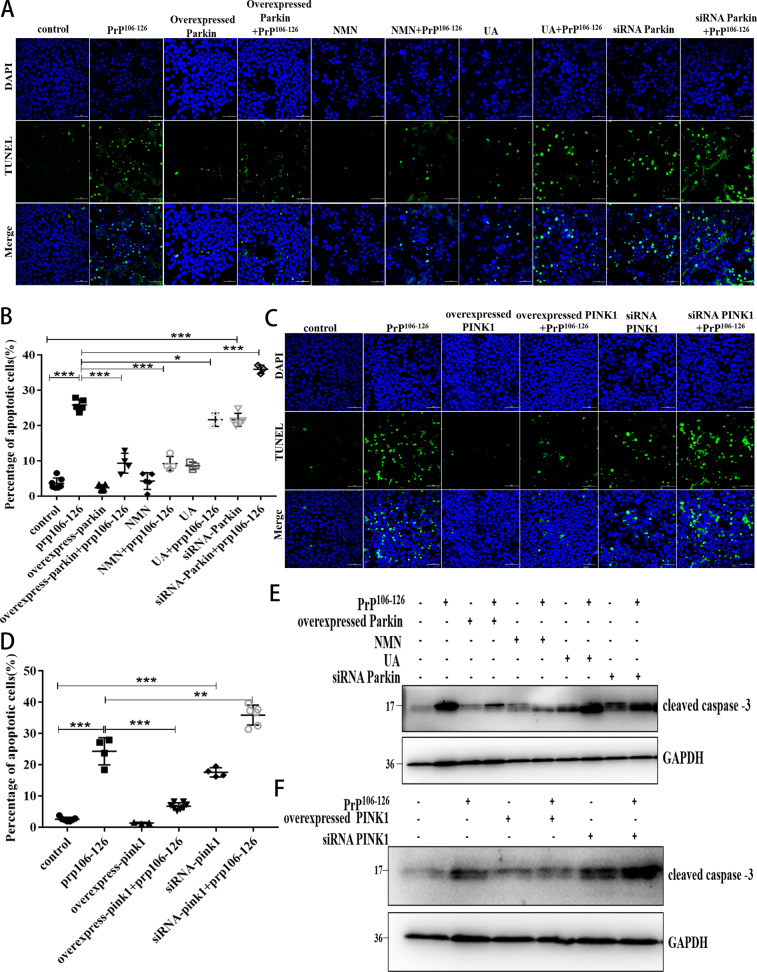
A persistent accumulation of damaged mitochondria is part of prion disease pathogenesis. Normally, damaged mitochondria are cleared via a major pathway that involves the E3 ubiquitin ligase parkin and PTEN-induced kinase 1 (PINK1) that together initiate mitophagy, recognize and eliminate damaged mitochondria. However, the precise mechanisms underlying mitophagy in prion disease remain largely unknown. Using prion disease cell models, we observed PINK1-parkin-mediated mitophagy deficiency in which parkin depletion aggravated blocked mitochondrial colocalization with LC3-II-labeled autophagosomes, and significantly increased mitochondrial protein levels, which led to inhibited mitophagy. Parkin overexpression directly induced LC3-II colocalization with mitochondria and alleviated defective mitophagy. Moreover, parkin-mediated mitophagy was dependent on PINK1, since PINK1 depletion blocked mitochondrial Parkin recruitment and reduced optineurin and LC3-II proteins levels, thus inhibiting mitophagy. PINK1 overexpression induced parkin recruitment to the mitochondria, which then stimulated mitophagy. In addition, overexpressed parkin and PINK1 also protected neurons from apoptosis. Furthermore, we found that supplementation with two mitophagy-inducing agents, nicotinamide mononucleotide (NMN) and urolithin A (UA), significantly stimulated PINK1-parkin-mediated mitophagy. However, compared with NMN, UA could not alleviate prion-induced mitochondrial fragmentation and dysfunction, and neuronal apoptosis. These findings show that PINK1-parkin-mediated mitophagy defects lead to an accumulation of damaged mitochondria, thus suggesting that interventions that stimulate mitophagy may be potential therapeutic targets for prion diseases.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Hur K, Kim JI, Choi SI, Choi EK, Carp RI, Kim YS. The pathogenic mechanisms of prion diseases. Mech Ageing Dev. 2002;123:1637–47. - PubMed
-
- Prusiner SB. Molecular biology and pathogenesis of prion diseases. Trends Biochem Sci. 1996;21:482–7. - PubMed
-
- Gu Y, Fujioka H, Mishra RS, Li R, Singh N. Prion peptide 106-126 modulates the aggregation of cellular prion protein and induces the synthesis of potentially neurotoxic transmembrane PrP. J Biol Chem. 2002;277:2275–86. - PubMed
-
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, et al. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–6. - PubMed
-
- Thellung S, Florio T, Villa V, Corsaro A, Arena S, Amico C, et al. Apoptotic cell death and impairment of L-type voltage-sensitive calcium channel activity in rat cerebellar granule cells treated with the prion protein fragment 106-126. Neurobiol Dis. 2000;7:299–309. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
