

Across-population genomic prediction in grapevine opens up promising prospects for breeding
- PMID: 35184162
- PMCID: PMC9070645
- DOI: 10.1093/hr/uhac041
Across-population genomic prediction in grapevine opens up promising prospects for breeding
Abstract
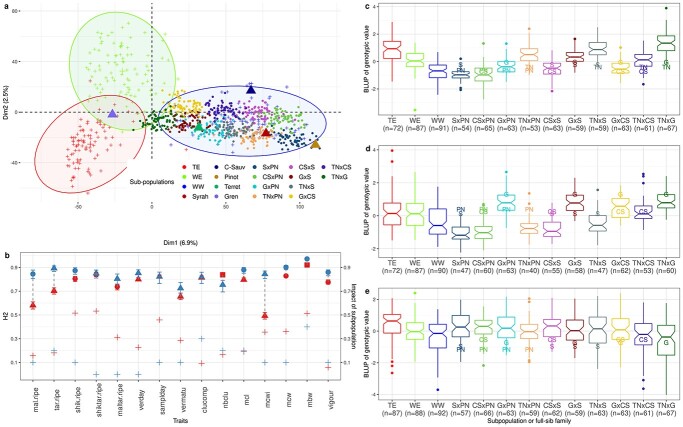
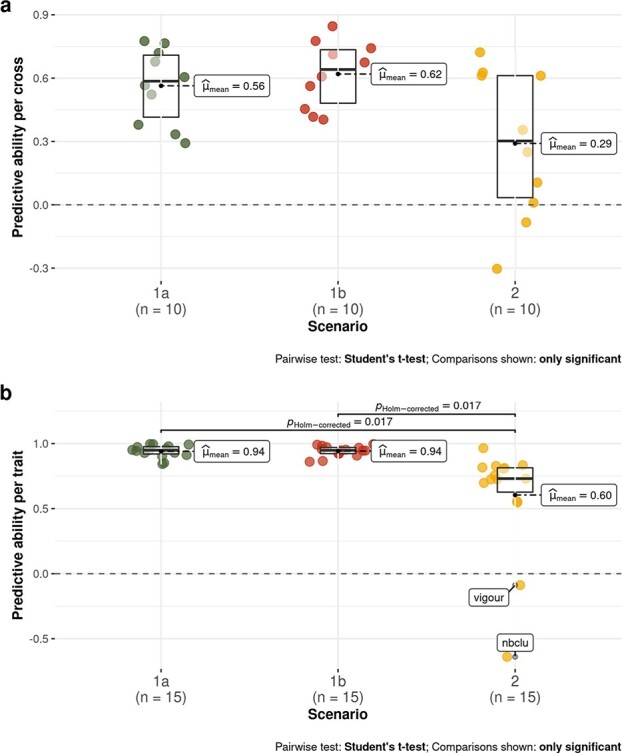
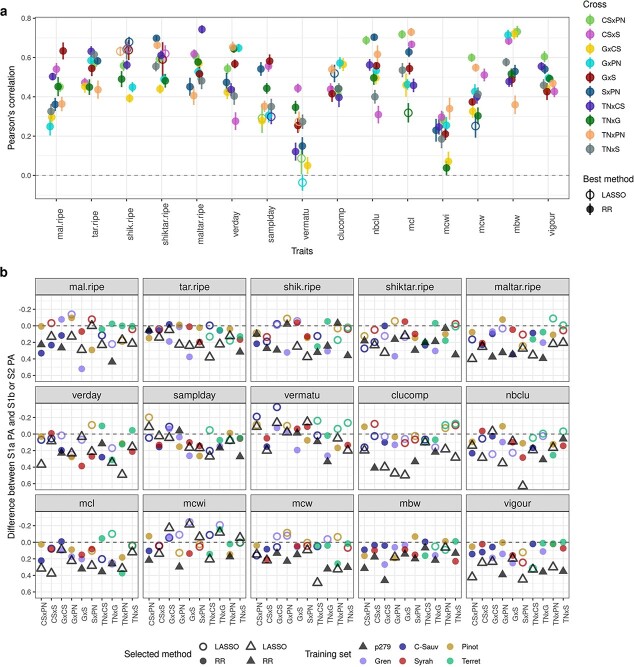
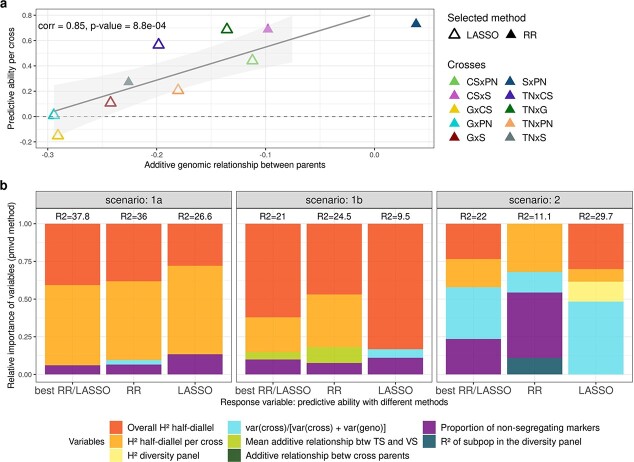
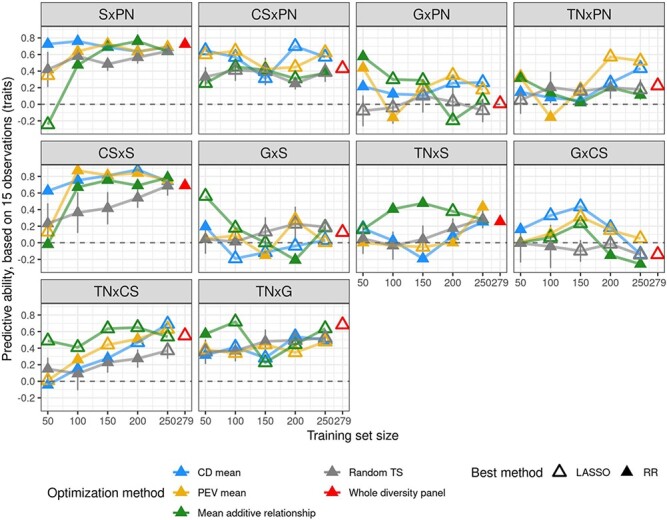
Crop breeding involves two selection steps: choosing progenitors and selecting individuals within progenies. Genomic prediction, based on genome-wide marker estimation of genetic values, could facilitate these steps. However, its potential usefulness in grapevine (Vitis vinifera L.) has only been evaluated in non-breeding contexts mainly through cross-validation within a single population. We tested across-population genomic prediction in a more realistic breeding configuration, from a diversity panel to ten bi-parental crosses connected within a half-diallel mating design. Prediction quality was evaluated over 15 traits of interest (related to yield, berry composition, phenology and vigour), for both the average genetic value of each cross (cross mean) and the genetic values of individuals within each cross (individual values). Genomic prediction in these conditions was found useful: for cross mean, average per-trait predictive ability was 0.6, while per-cross predictive ability was halved on average, but reached a maximum of 0.7. Mean predictive ability for individual values within crosses was 0.26, about half the within-half-diallel value taken as a reference. For some traits and/or crosses, these across-population predictive ability values are promising for implementing genomic selection in grapevine breeding. This study also provided key insights on variables affecting predictive ability. Per-cross predictive ability was well predicted by genetic distance between parents and when this predictive ability was below 0.6, it was improved by training set optimization. For individual values, predictive ability mostly depended on trait-related variables (magnitude of the cross effect and heritability). These results will greatly help designing grapevine breeding programs assisted by genomic prediction.
Keywords: across-population; diversity panel; genomic prediction; grapevine; half-diallel; multi-parental population.
© The Author(s) 2022. Published by Oxford University Press. All rights reserved.
Figures

References
-
- Heffner EL, Sorrells ME, Jannink J-L. Genomic selection for crop improvement. Crop Sci. 2009;157:1819–29.
-
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. Pearson: Prentice Hall; 2009.
-
- Bernardo R. Genomewide selection of parental Inbreds: classes of loci and virtual Biparental populations. Crop Sci. 2014;54:2586–95.
LinkOut - more resources
Full Text Sources
