

Crosstalk Between the NLRP3 Inflammasome/ASC Speck and Amyloid Protein Aggregates Drives Disease Progression in Alzheimer's and Parkinson's Disease
- PMID: 35185469
- PMCID: PMC8850380
- DOI: 10.3389/fnmol.2022.805169
Crosstalk Between the NLRP3 Inflammasome/ASC Speck and Amyloid Protein Aggregates Drives Disease Progression in Alzheimer's and Parkinson's Disease
Abstract
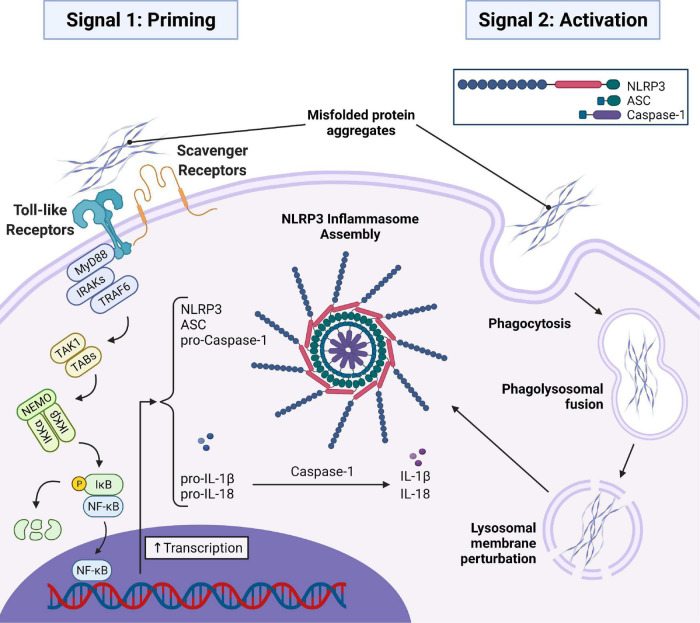
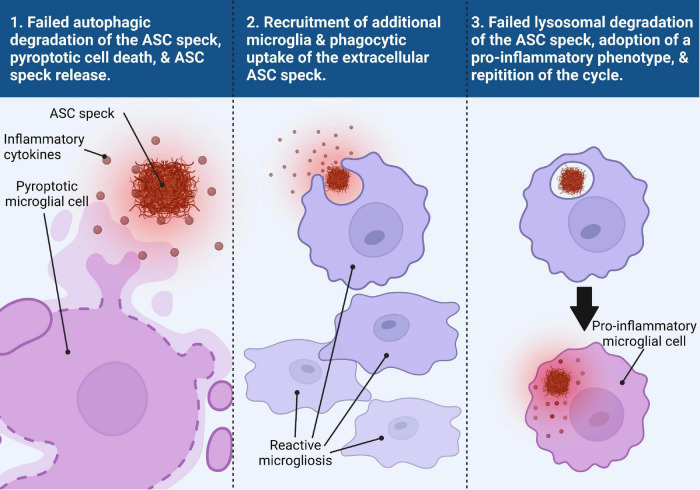
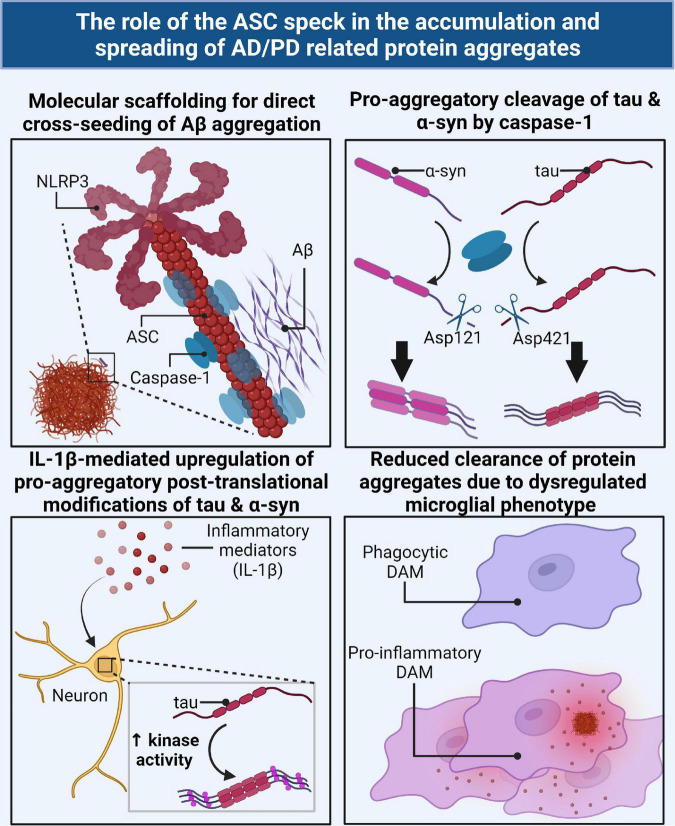
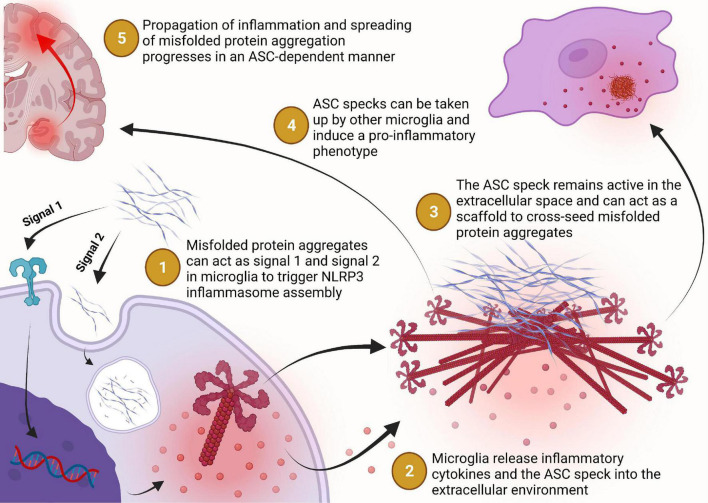
Two key pathological hallmarks of neurodegenerative diseases, including Alzheimer's disease (AD) and Parkinson's disease (PD), are the accumulation of misfolded protein aggregates and the chronic progressive neuroinflammation that they trigger. Numerous original research and reviews have provided a comprehensive understanding of how aggregated proteins (amyloid β, pathological tau, and α-synuclein) contribute to the disease, including driving sterile inflammation, in part, through the aggregation of multi-protein inflammasome complexes and the ASC speck [composed of NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3), Apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain (ASC), and inflammatory caspase-1] involved in innate immunity. Here, we provide a unique perspective on the crosstalk between the aggregation-prone proteins involved in AD/PD and the multi-protein inflammasome complex/ASC speck that fuels feed-forward exacerbation of each other, driving neurodegeneration. Failed turnover of protein aggregates (both AD/PD related aggregates and the ASC speck) by protein degradation pathways, prionoid propagation of inflammation by the ASC speck, cross-seeding of protein aggregation by the ASC speck, and pro-aggregatory cleavage of proteins by caspase-1 are some of the mechanisms that exacerbate disease progression. We also review studies that provide this causal framework and highlight how the ASC speck serves as a platform for the propagation and spreading of inflammation and protein aggregation that drives AD and PD.
Keywords: ASC; Alzheimer’s disease; NLRP3; Parkinson’s disease; autophagy-lysosomal degradation; inflammasomes; neuroinflammation; protein aggregation.
Copyright © 2022 Hulse and Bhaskar.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous