

Modes of Brain Cell Death Following Intracerebral Hemorrhage
- PMID: 35185473
- PMCID: PMC8851202
- DOI: 10.3389/fncel.2022.799753
Modes of Brain Cell Death Following Intracerebral Hemorrhage
Abstract
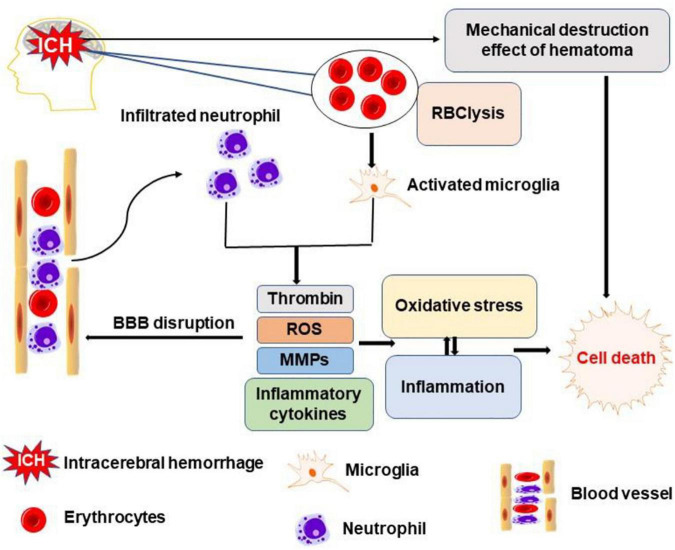
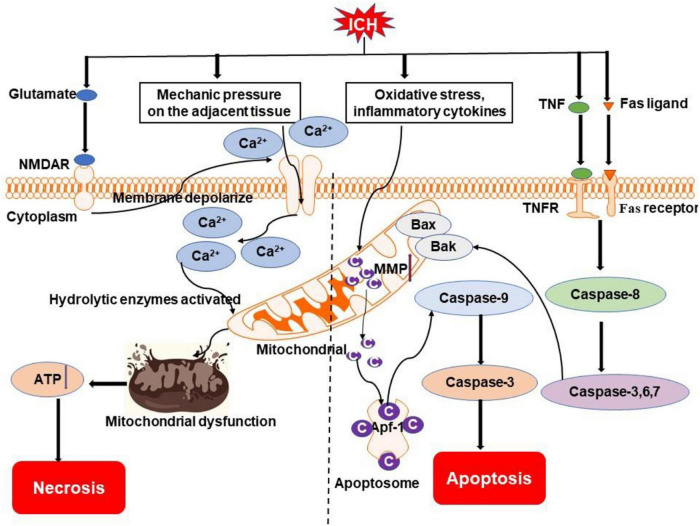
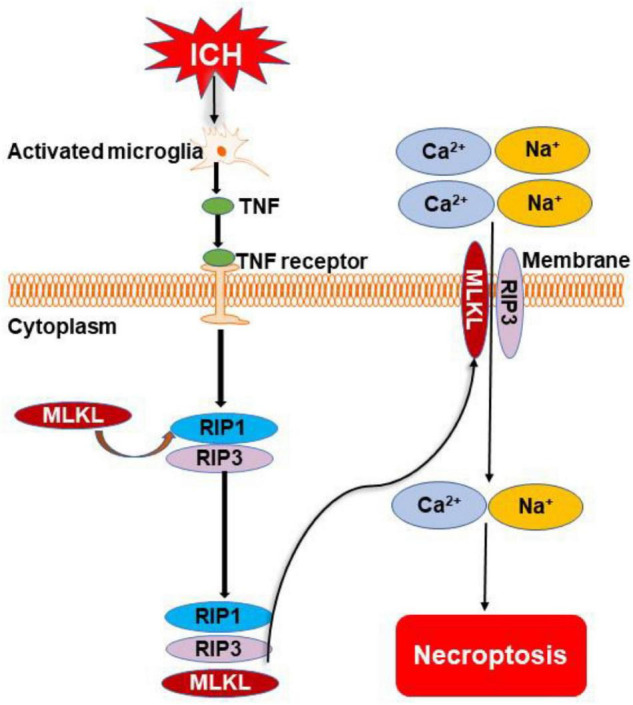
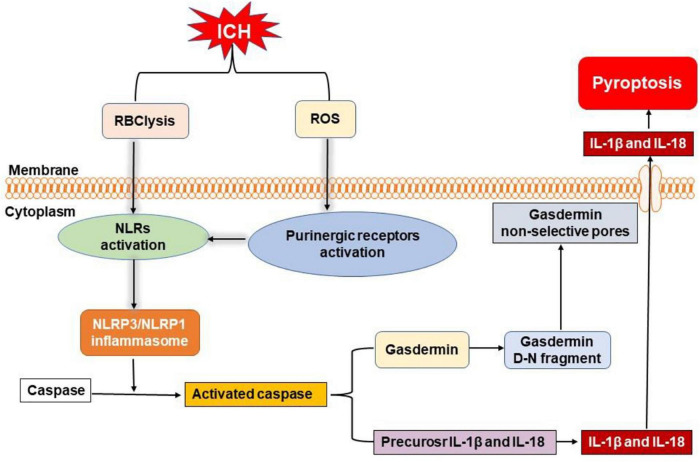
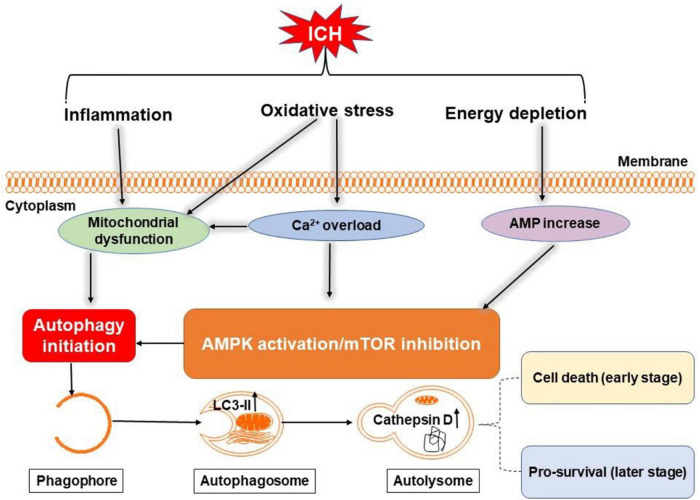
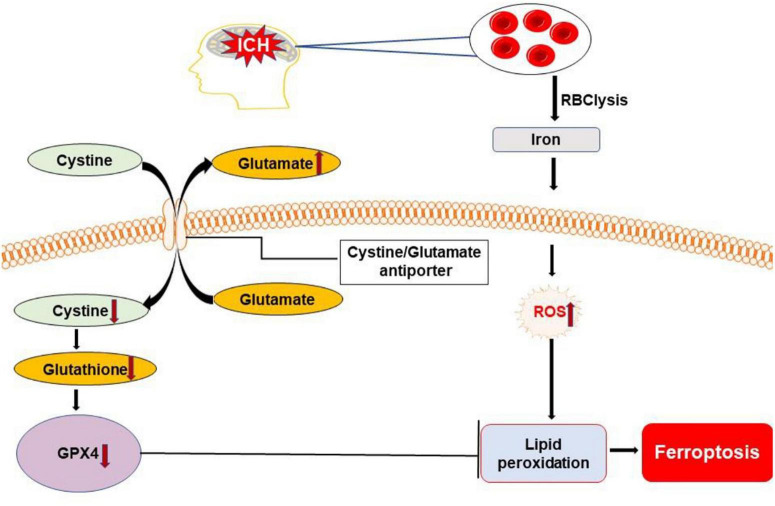
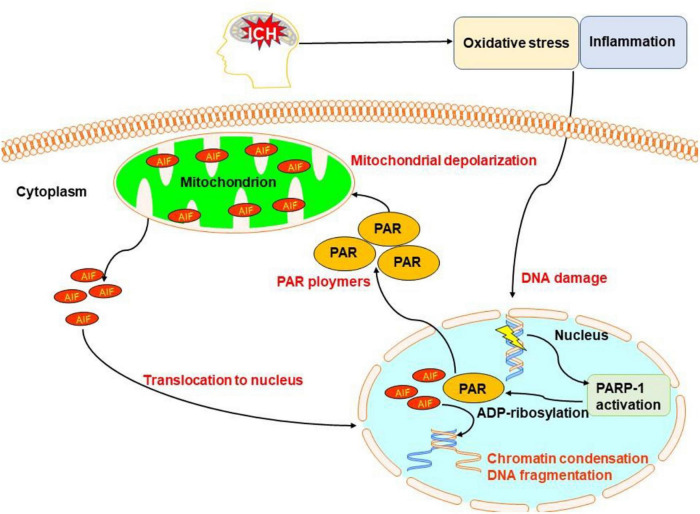
Intracerebral hemorrhage (ICH) is a devastating form of stroke with high rates of mortality and morbidity. It induces cell death that is responsible for neurological deficits postinjury. There are no therapies that effectively mitigate cell death to treat ICH. This review aims to summarize our knowledge of ICH-induced cell death with a focus on apoptosis and necrosis. We also discuss the involvement of ICH in recently described modes of cell death including necroptosis, pyroptosis, ferroptosis, autophagy, and parthanatos. We summarize treatment strategies to mitigate brain injury based on particular cell death pathways after ICH.
Keywords: apoptosis; autophagy; cell death; ferroptosis; intracerebral hemorrhage; necrosis; parthanatos.
Copyright © 2022 Zhang, Khan, Liu, Zhang, Li, Wu, Tang, Xue and Yong.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources