

Whole-genome analysis of SARS-CoV-2 in a 2020 infection cluster in a nursing home of Southern Italy
- PMID: 35189404
- PMCID: PMC8855624
- DOI: 10.1016/j.meegid.2022.105253
Whole-genome analysis of SARS-CoV-2 in a 2020 infection cluster in a nursing home of Southern Italy
Abstract
Background: Nursing homes have represented important hotspots of viral spread during the initial wave of COVID-19 pandemics. The proximity of patients inside nursing homes allows investigate the dynamics of viral transmission, which may help understand SARS-Cov2 biology and spread.
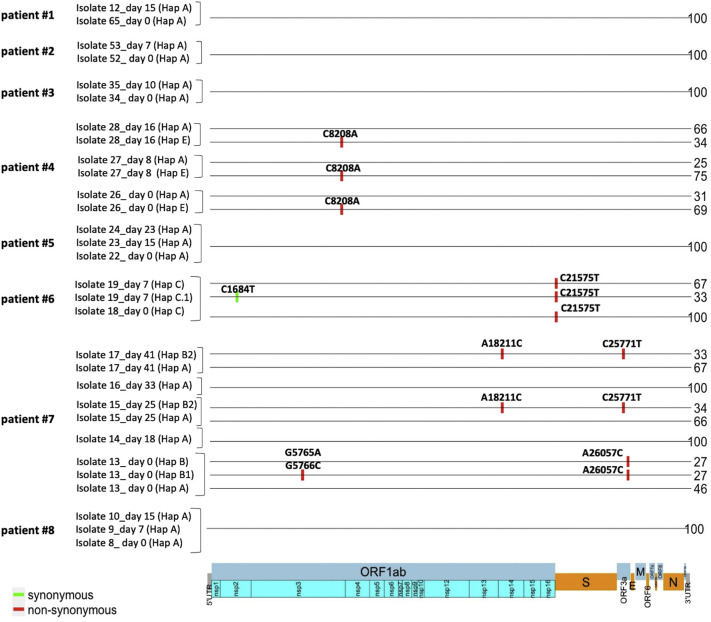
Methods: SARS-CoV-2 viral genomes obtained from 46 patients infected in an outbreak inside a nursing home in Calabria region (South Italy) were analyzed by Next Generation Sequencing. We also investigated the evolution of viral genomes in 8 patients for which multiple swabs were available. Phylogenetic analysis and haplotype reconstruction were carried out with IQ-TREE software and RegressHaplo tool, respectively.
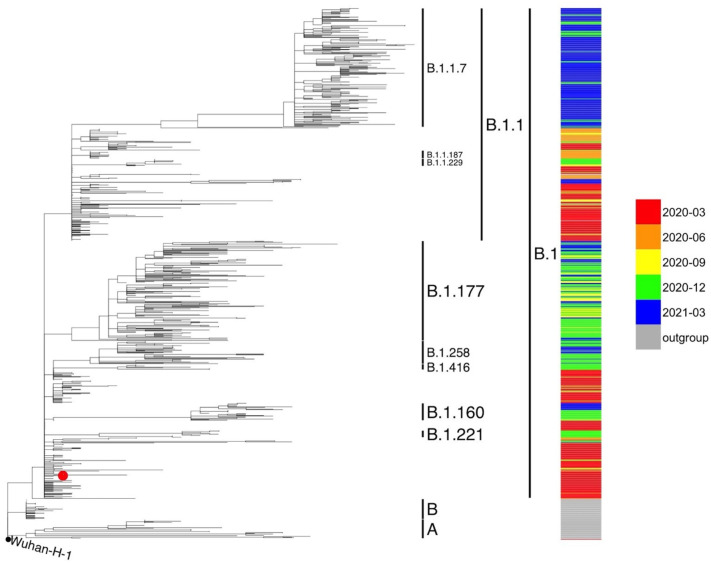
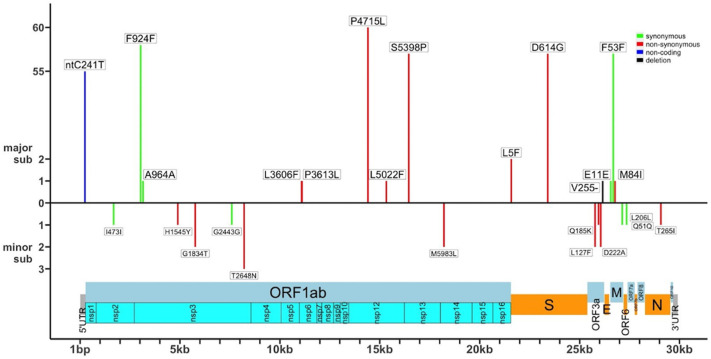
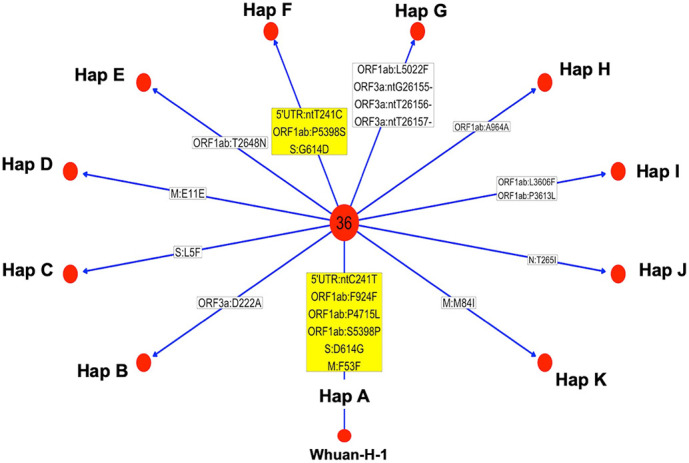
Results: All viral strains isolated from patients infected in the nursing home were classified as B.1 lineage, clade G. Overall, 14 major single nucleotide variations (SNVs) (frequency > 80%) and 12 minor SNVs (frequency comprised between 20% and 80%) were identified with reference to the Wuhan-H-1 sequence (NC_045512.2). All patients presented the same 6 major SNVs: D614G in the S gene; P4715L, ntC3037T (F924F) and S5398P in Orf1ab gene; ntC26681T (F53F) in the M gene; and ntC241T in the non-coding UTR region. However, haplotype reconstruction identified a founder haplotype (Hap A) in 36 patients carrying only the 6 common SNVs indicated above, and 10 other haplotypes (Hap BK) derived from Hap A in the remaining 10 patients. Notably, no significant association between a specific viral haplotype and clinical parameters was found.
Conclusion: The predominant viral strain responsible for the infection in a nursing home in Calabria was the B.1 lineage (clade G). Viral genomes were classified into 11 haplotypes (Hap A in 36 patients, Hap BK in the remaining patients).
Keywords: COVID-19; Haplotypes; NGS; SARS-CoV-2.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
