

Systematic evaluation of computational tools to predict the effects of mutations on protein stability in the absence of experimental structures
- PMID: 35189634
- PMCID: PMC9155634
- DOI: 10.1093/bib/bbac025
Systematic evaluation of computational tools to predict the effects of mutations on protein stability in the absence of experimental structures
Abstract
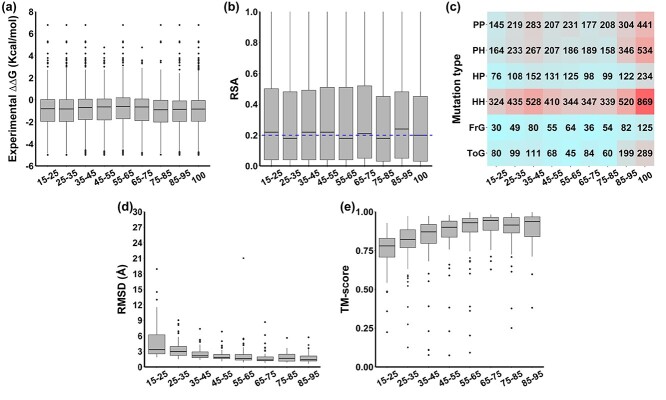
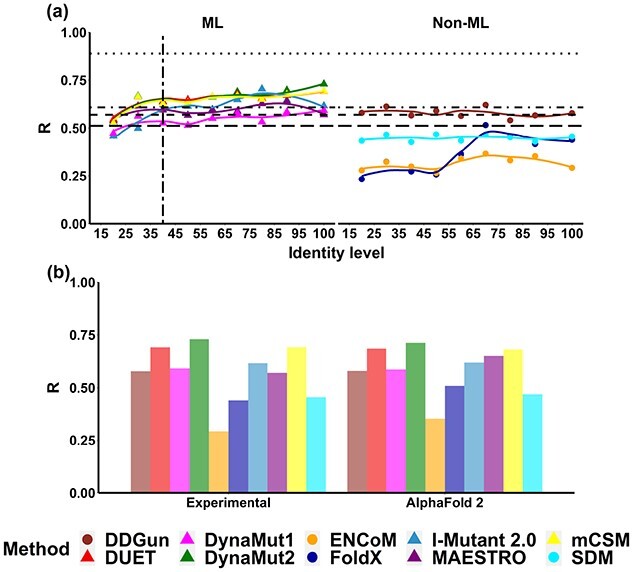
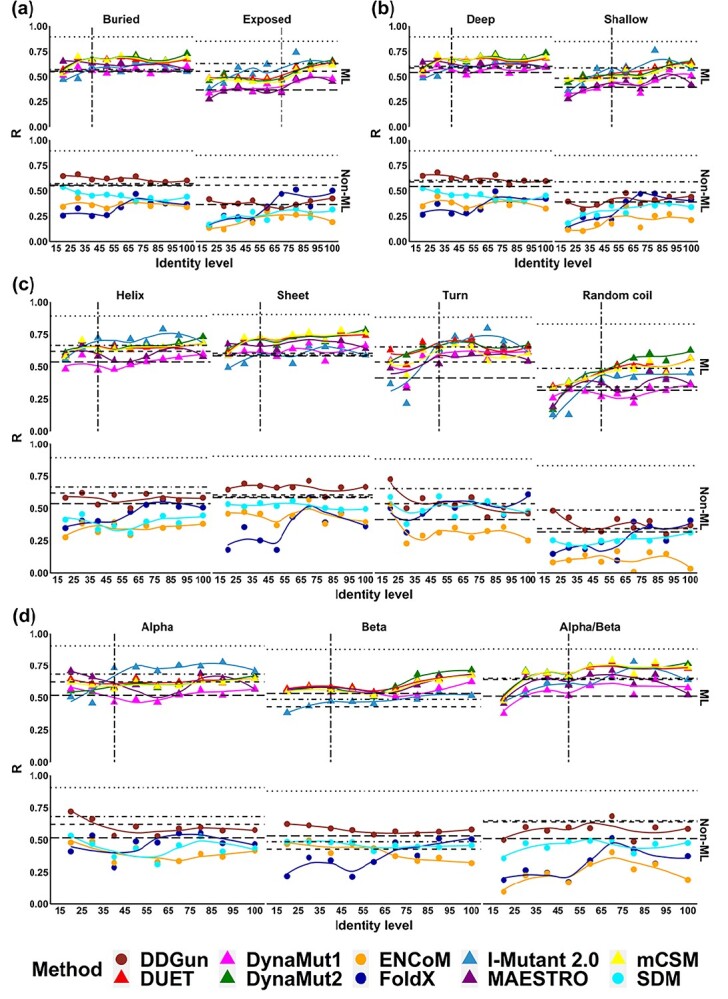
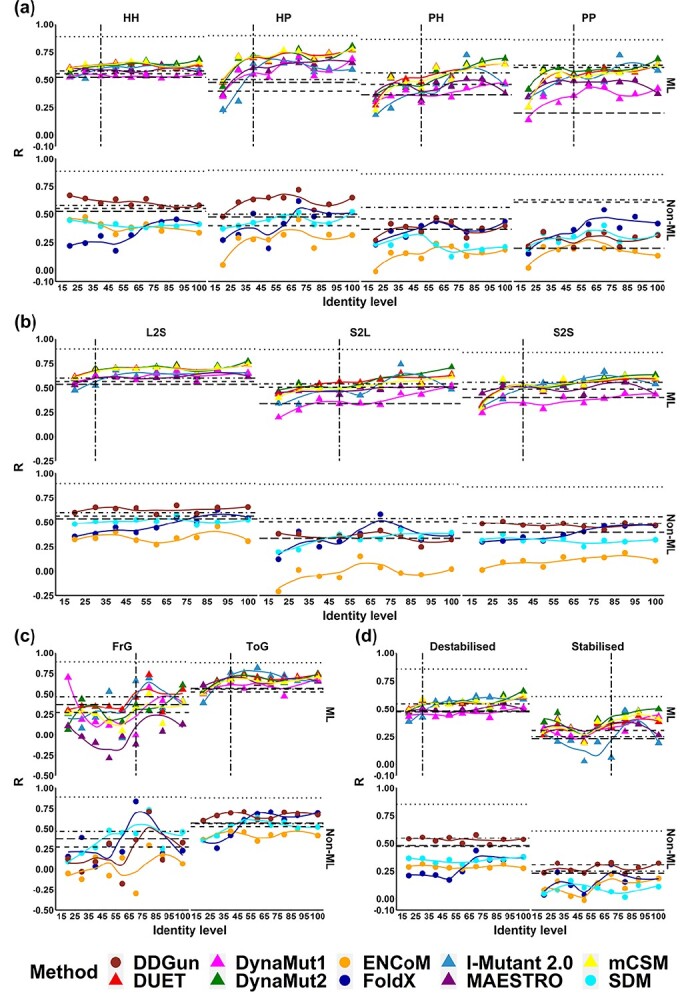
Changes in protein sequence can have dramatic effects on how proteins fold, their stability and dynamics. Over the last 20 years, pioneering methods have been developed to try to estimate the effects of missense mutations on protein stability, leveraging growing availability of protein 3D structures. These, however, have been developed and validated using experimentally derived structures and biophysical measurements. A large proportion of protein structures remain to be experimentally elucidated and, while many studies have based their conclusions on predictions made using homology models, there has been no systematic evaluation of the reliability of these tools in the absence of experimental structural data. We have, therefore, systematically investigated the performance and robustness of ten widely used structural methods when presented with homology models built using templates at a range of sequence identity levels (from 15% to 95%) and contrasted performance with sequence-based tools, as a baseline. We found there is indeed performance deterioration on homology models built using templates with sequence identity below 40%, where sequence-based tools might become preferable. This was most marked for mutations in solvent exposed residues and stabilizing mutations. As structure prediction tools improve, the reliability of these predictors is expected to follow, however we strongly suggest that these factors should be taken into consideration when interpreting results from structure-based predictors of mutation effects on protein stability.
Keywords: AlphaFold2; homology modelling; mutation effects on protein stability; performance evaluation.
© The Author(s) 2022. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Comment in
-
Influence of Model Structures on Predictors of Protein Stability Changes from Single-Point Mutations.Genes (Basel). 2023 Dec 17;14(12):2228. doi: 10.3390/genes14122228. Genes (Basel). 2023. PMID: 38137050 Free PMC article.
References
-
- Jafri M, Wake NC, Ascher DB, et al. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell carcinoma. Cancer Discov 2015;5:723–9. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
