

The nephropathy of sickle cell trait and sickle cell disease
- PMID: 35190716
- PMCID: PMC9832386
- DOI: 10.1038/s41581-022-00540-9
The nephropathy of sickle cell trait and sickle cell disease
Abstract
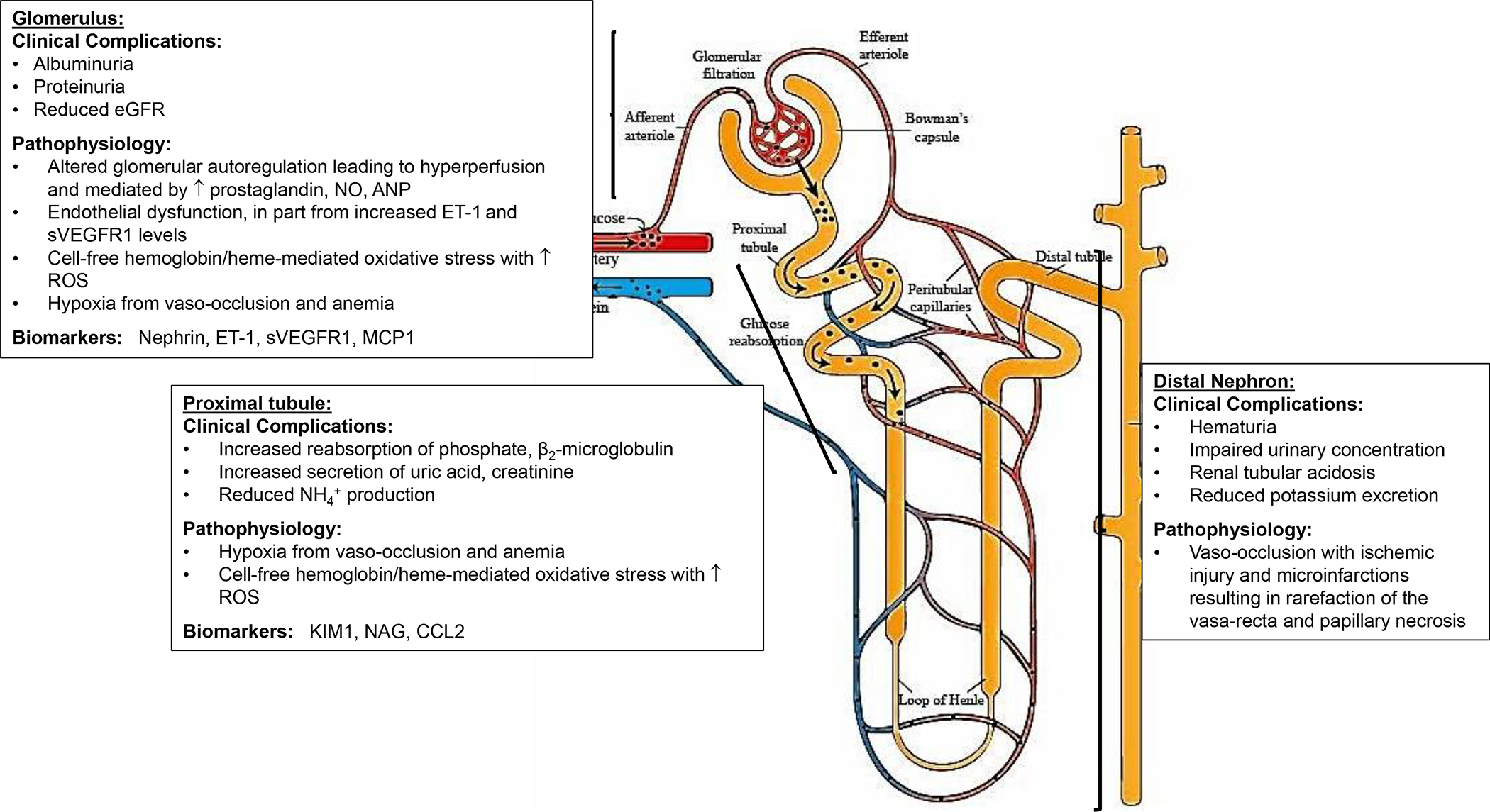
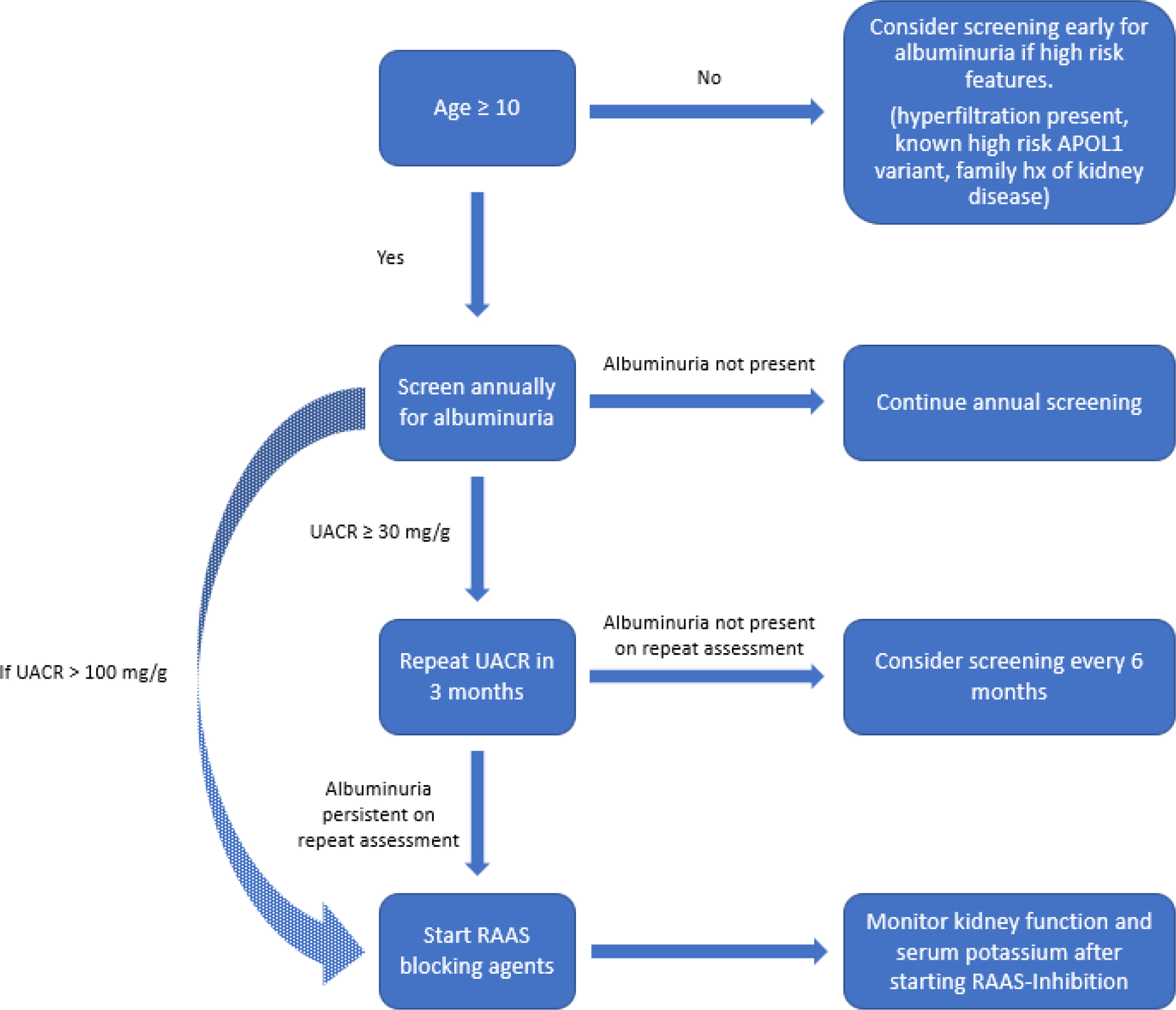
Sickle cell syndromes, including sickle cell disease (SCD) and sickle cell trait, are associated with multiple kidney abnormalities. Young patients with SCD have elevated effective renal plasma flow and glomerular filtration rates, which decrease to normal ranges in young adulthood and subnormal levels with advancing age. The pathophysiology of SCD-related nephropathy is multifactorial - oxidative stress, hyperfiltration and glomerular hypertension are all contributing factors. Albuminuria, which is an early clinical manifestation of glomerular damage, is common in individuals with SCD. Kidney function declines more rapidly in individuals with SCD than in those with sickle cell trait or in healthy individuals. Multiple genetic modifiers, including APOL1, HMOX1, HBA1 and HBA2 variants are also implicated in the development and progression of SCD-related nephropathy. Chronic kidney disease and rapid decline in estimated glomerular filtration rate are associated with increased mortality in adults with SCD. Renin-angiotensin-aldosterone system inhibitors are the standard of care treatment for albuminuria in SCD, despite a lack of controlled studies demonstrating their long-term efficacy. Multiple studies of novel therapeutic agents are ongoing, and patients with SCD and kidney failure should be evaluated for kidney transplantation. Given the high prevalence and severe consequences of kidney disease, additional studies are needed to elucidate the pathophysiology, natural history and treatment of SCD-related nephropathy.
© 2022. Springer Nature Limited.
Conflict of interest statement
Competing interests
K.I.A. has received research funding from Novartis and Global Blood Therapeutics, served on advisory boards for Novartis, Global Blood Therapeutics, Novo Nordisk, Editas Medicine, Forma Therapeutics and Agios Pharmaceuticals, and as a consultant for Roche. S.L.S. receives research funding support from Novartis, Pfizer and Global Blood Therapeutics, and served on advisory boards for Novartis and Global Blood Therapeutics. V.K.D. has served on advisory boards for Novartis, Bayer and Travere.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
