

Sequence determinants of human gene regulatory elements
- PMID: 35190730
- PMCID: PMC8920891
- DOI: 10.1038/s41588-021-01009-4
Sequence determinants of human gene regulatory elements
Abstract
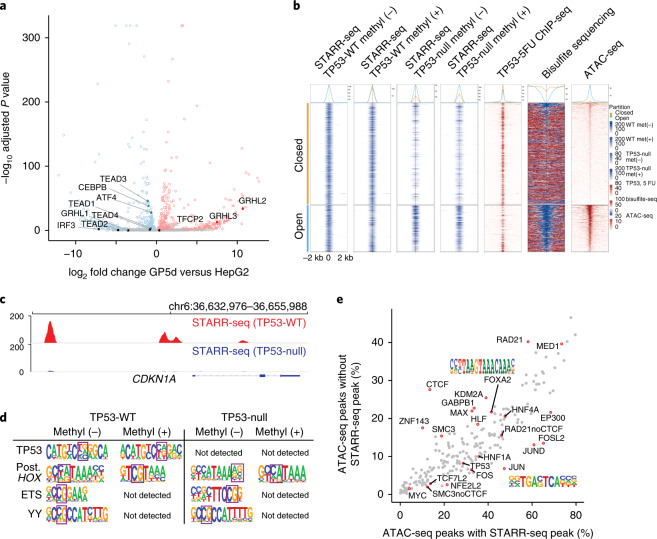
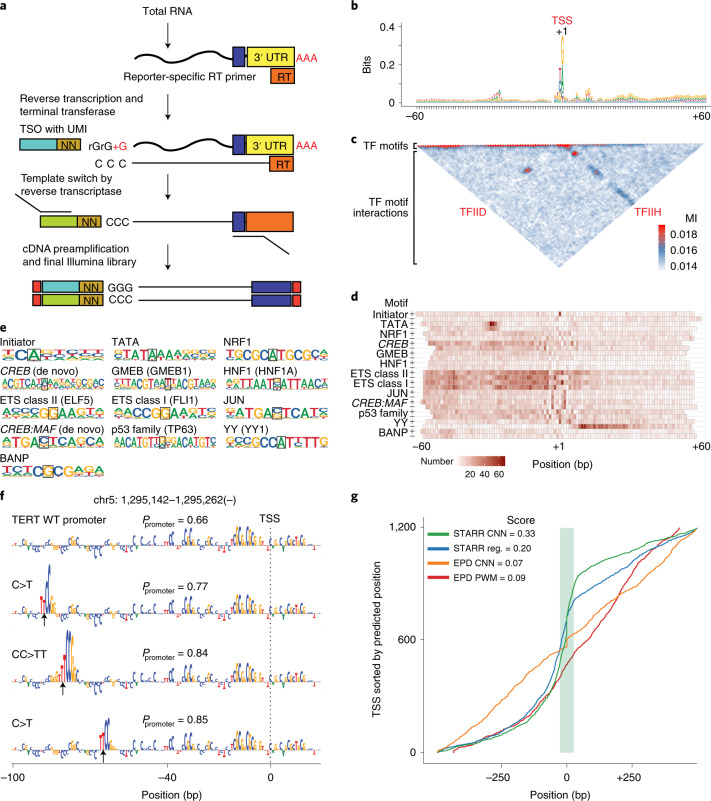
DNA can determine where and when genes are expressed, but the full set of sequence determinants that control gene expression is unknown. Here, we measured the transcriptional activity of DNA sequences that represent an ~100 times larger sequence space than the human genome using massively parallel reporter assays (MPRAs). Machine learning models revealed that transcription factors (TFs) generally act in an additive manner with weak grammar and that most enhancers increase expression from a promoter by a mechanism that does not appear to involve specific TF-TF interactions. The enhancers themselves can be classified into three types: classical, closed chromatin and chromatin dependent. We also show that few TFs are strongly active in a cell, with most activities being similar between cell types. Individual TFs can have multiple gene regulatory activities, including chromatin opening and enhancing, promoting and determining transcription start site (TSS) activity, consistent with the view that the TF binding motif is the key atomic unit of gene expression.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures














References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
