

Assessing the landscape of STXBP1-related disorders in 534 individuals
- PMID: 35190816
- PMCID: PMC9166568
- DOI: 10.1093/brain/awab327
Assessing the landscape of STXBP1-related disorders in 534 individuals
Abstract
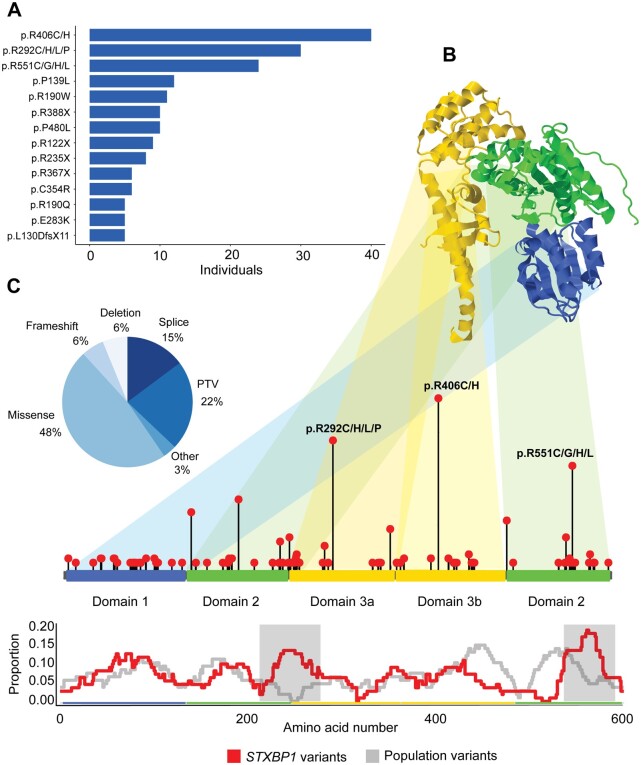
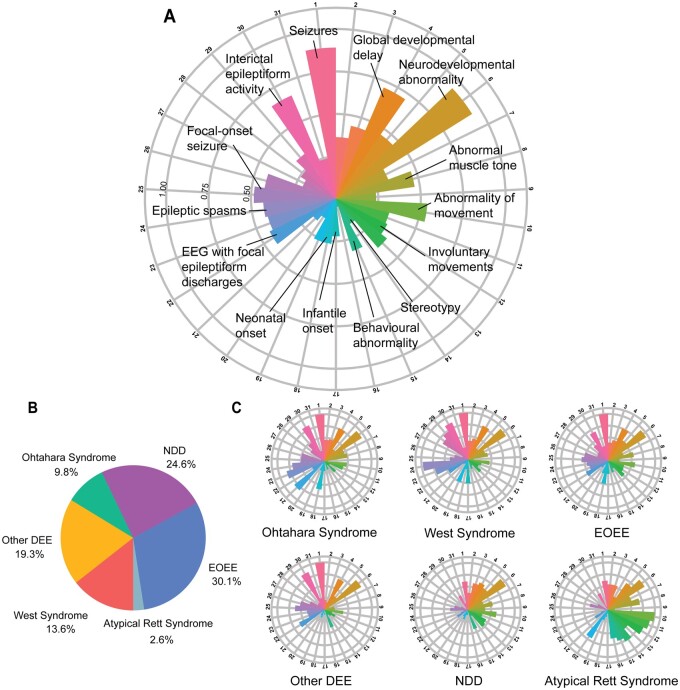
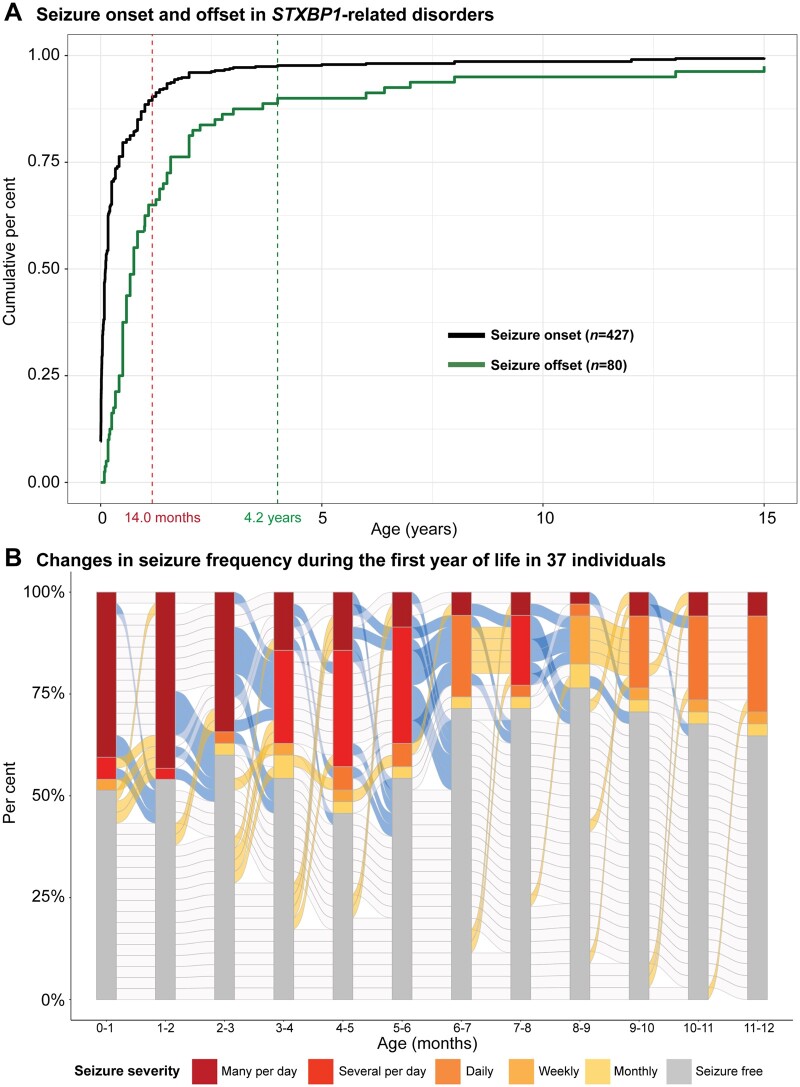
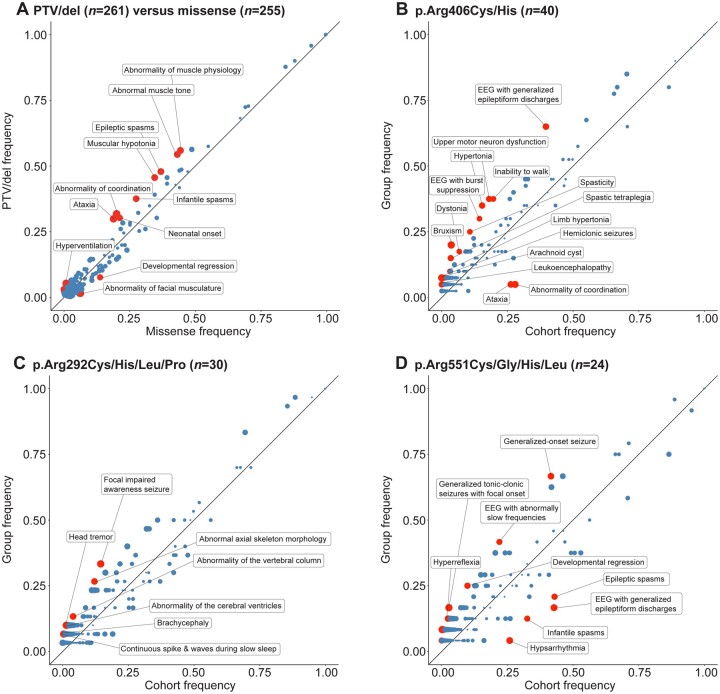
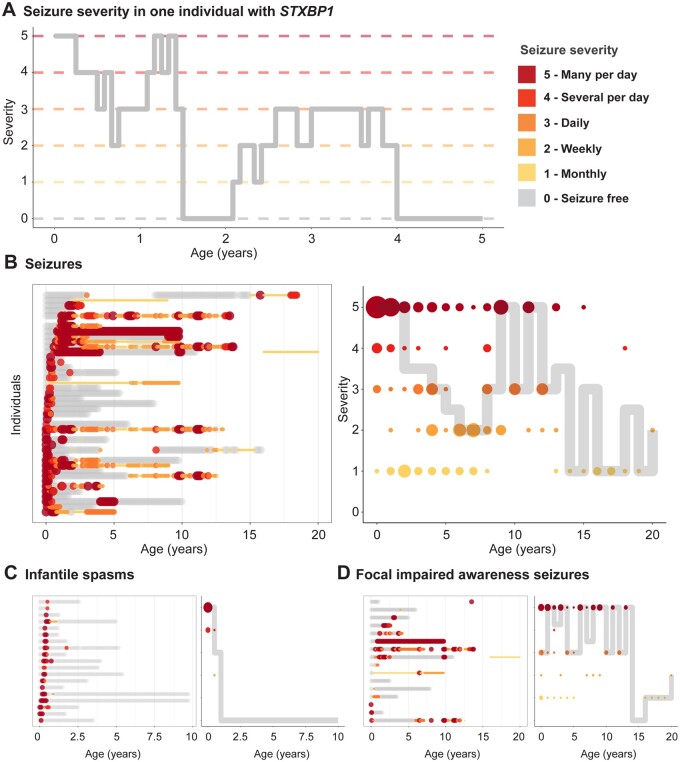
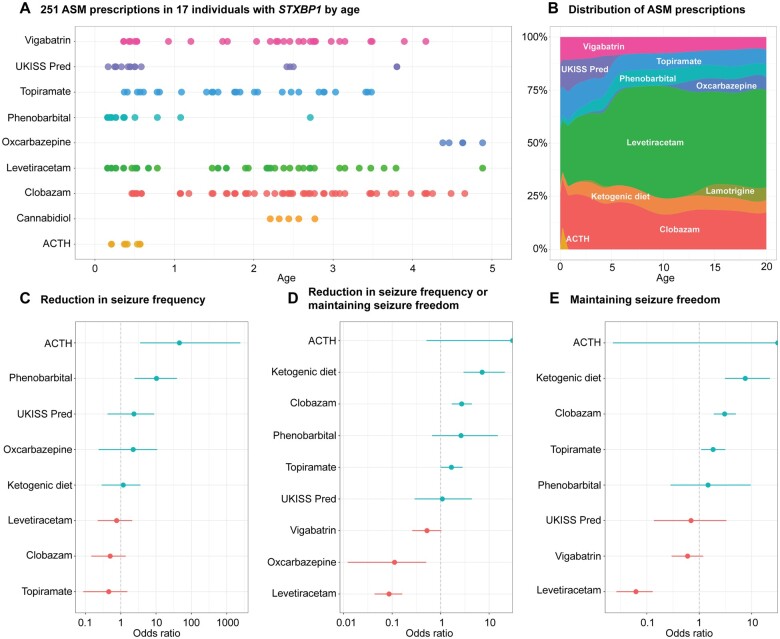
Disease-causing variants in STXBP1 are among the most common genetic causes of neurodevelopmental disorders. However, the phenotypic spectrum in STXBP1-related disorders is wide and clear correlations between variant type and clinical features have not been observed so far. Here, we harmonized clinical data across 534 individuals with STXBP1-related disorders and analysed 19 973 derived phenotypic terms, including phenotypes of 253 individuals previously unreported in the scientific literature. The overall phenotypic landscape in STXBP1-related disorders is characterized by neurodevelopmental abnormalities in 95% and seizures in 89% of individuals, including focal-onset seizures as the most common seizure type (47%). More than 88% of individuals with STXBP1-related disorders have seizure onset in the first year of life, including neonatal seizure onset in 47%. Individuals with protein-truncating variants and deletions in STXBP1 (n = 261) were almost twice as likely to present with West syndrome and were more phenotypically similar than expected by chance. Five genetic hotspots with recurrent variants were identified in more than 10 individuals, including p.Arg406Cys/His (n = 40), p.Arg292Cys/His/Leu/Pro (n = 30), p.Arg551Cys/Gly/His/Leu (n = 24), p.Pro139Leu (n = 12), and p.Arg190Trp (n = 11). None of the recurrent variants were significantly associated with distinct electroclinical syndromes, single phenotypic features, or showed overall clinical similarity, indicating that the baseline variability in STXBP1-related disorders is too high for discrete phenotypic subgroups to emerge. We then reconstructed the seizure history in 62 individuals with STXBP1-related disorders in detail, retrospectively assigning seizure type and seizure frequency monthly across 4433 time intervals, and retrieved 251 anti-seizure medication prescriptions from the electronic medical records. We demonstrate a dynamic pattern of seizure control and complex interplay with response to specific medications particularly in the first year of life when seizures in STXBP1-related disorders are the most prominent. Adrenocorticotropic hormone and phenobarbital were more likely to initially reduce seizure frequency in infantile spasms and focal seizures compared to other treatment options, while the ketogenic diet was most effective in maintaining seizure freedom. In summary, we demonstrate how the multidimensional spectrum of phenotypic features in STXBP1-related disorders can be assessed using a computational phenotype framework to facilitate the development of future precision-medicine approaches.
Keywords: STXBP1; Human Phenotype Ontology; developmental and epileptic encephalopathy; epilepsy; genetics.
© The Author(s) (2021). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
References
-
- Stamberger H, Nikanorova M, Willemsen MH, et al. STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology. 2016;86(10):954–962. - PubMed
-
- Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40(6):782–788. - PubMed
