

High-throughput identification and quantification of single bacterial cells in the microbiota
- PMID: 35194029
- PMCID: PMC8863893
- DOI: 10.1038/s41467-022-28426-1
High-throughput identification and quantification of single bacterial cells in the microbiota
Abstract
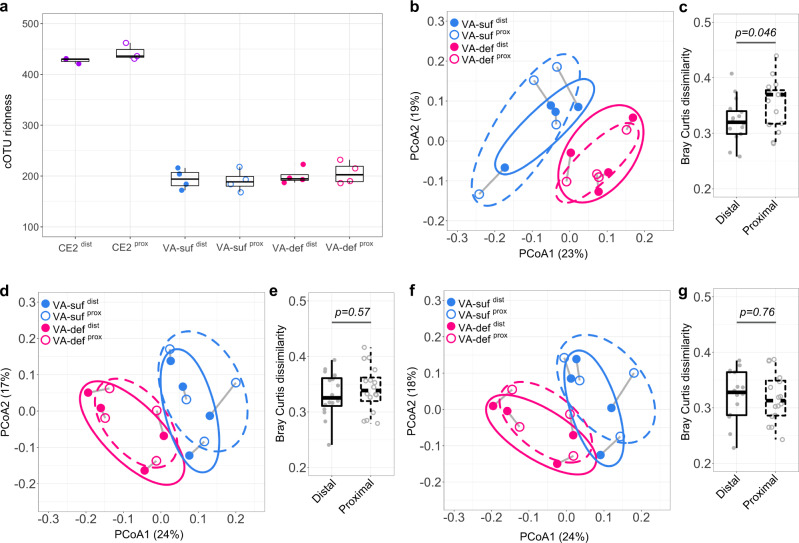
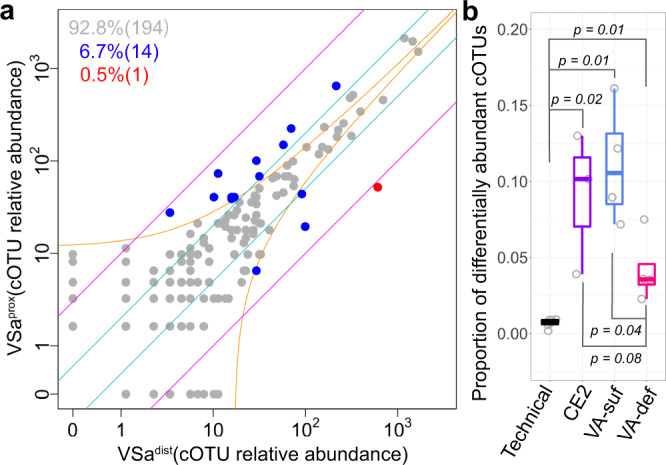
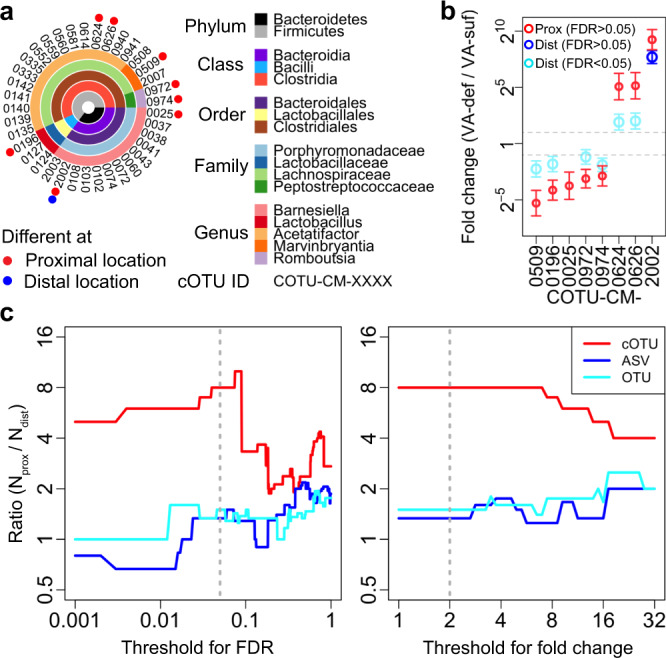
The bacterial microbiota works as a community that consists of many individual organisms, i.e., cells. To fully understand the function of bacterial microbiota, individual cells must be identified; however, it is difficult with current techniques. Here, we develop a method, Barcoding Bacteria for Identification and Quantification (BarBIQ), which classifies single bacterial cells into taxa-named herein cell-based operational taxonomy units (cOTUs)-based on cellularly barcoded 16S rRNA sequences with single-base accuracy, and quantifies the cell number for each cOTU in the microbiota in a high-throughput manner. We apply BarBIQ to murine cecal microbiotas and quantify in total 3.4 × 105 bacterial cells containing 810 cOTUs. Interestingly, we find location-dependent global differences in the cecal microbiota depending on the dietary vitamin A deficiency, and more differentially abundant cOTUs at the proximal location than the distal location. Importantly, these location differences are not clearly shown by conventional 16S rRNA gene-amplicon sequencing methods, which quantify the 16S rRNA genes, not the cells. Thus, BarBIQ enables microbiota characterization with the identification and quantification of individual constituent bacteria, which is a cornerstone for microbiota studies.
© 2022. The Author(s).
Conflict of interest statement
K.S., J.J., and R.Y. are co-inventors on a patent application based on this work filed by RIKEN. The other authors declare no competing interests.
Figures
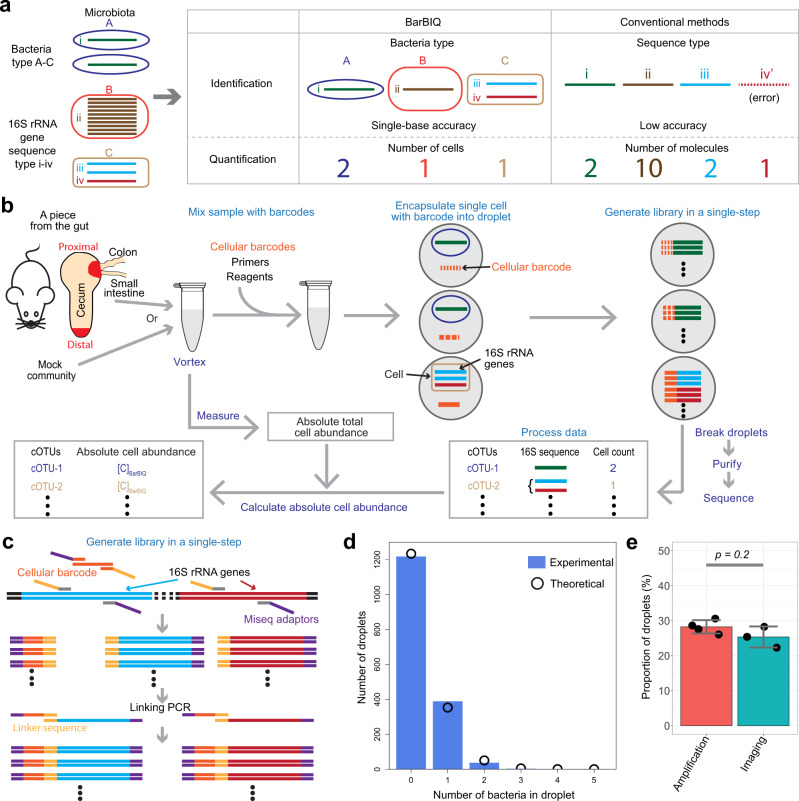
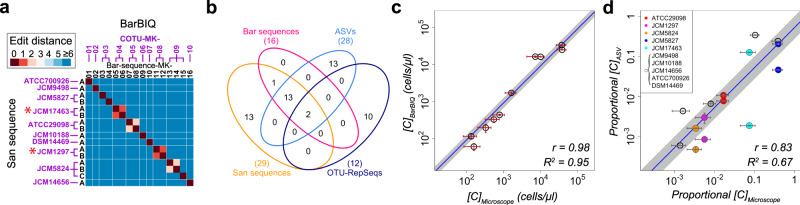
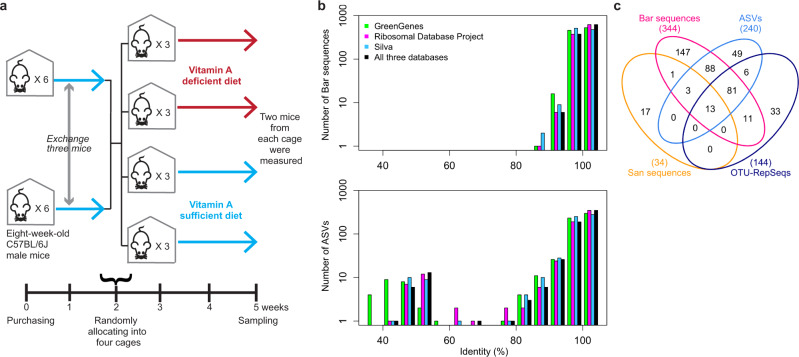
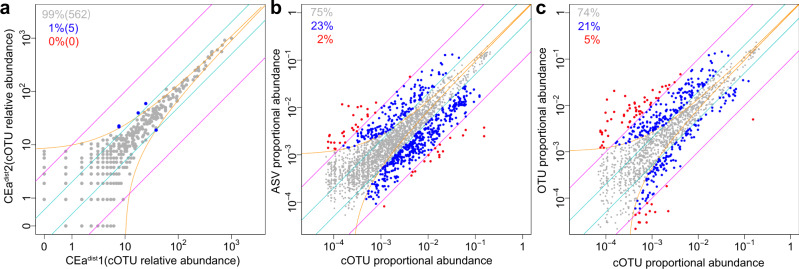
References
-
- Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164:337–340. - PubMed
-
- Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010;10:159–169. - PubMed
-
- Vandeputte D, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551:507–511. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
