

Diversity of Pseudomonas aeruginosa Temperate Phages
- PMID: 35196122
- PMCID: PMC8865926
- DOI: 10.1128/msphere.01015-21
Diversity of Pseudomonas aeruginosa Temperate Phages
Abstract
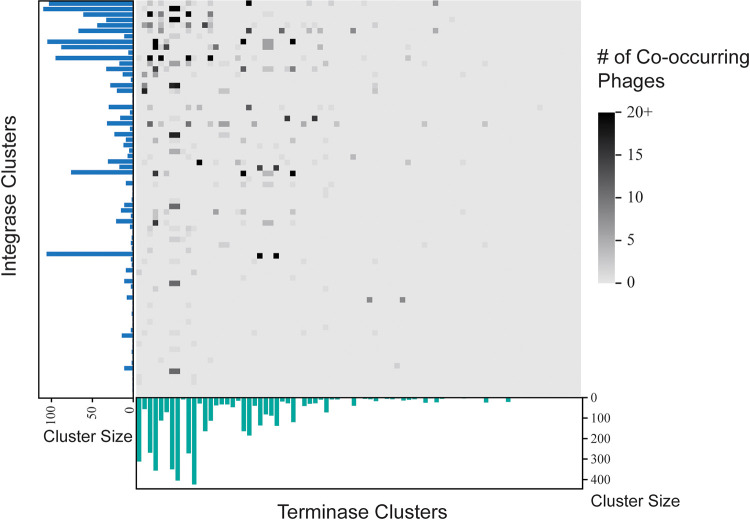
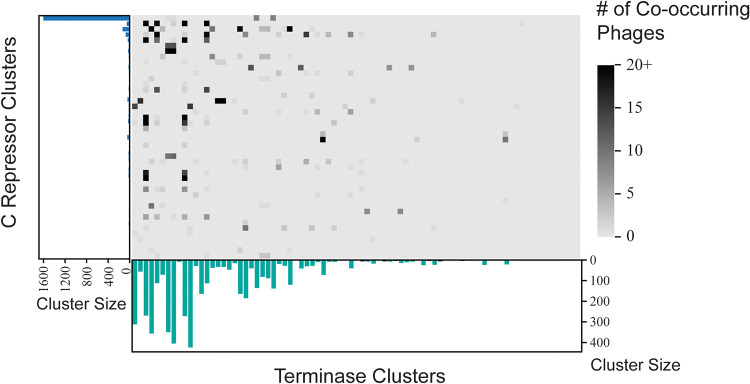
Modern sequencing technologies have provided insight into the genetic diversity of numerous species, including the human pathogen Pseudomonas aeruginosa. Bacterial genomes often harbor bacteriophage genomes (prophages), which can account for upwards of 20% of the genome. Prior studies have found P. aeruginosa prophages that contribute to their host's pathogenicity and fitness. These advantages come in many different forms, including the production of toxins, promotion of biofilm formation, and displacement of other P. aeruginosa strains. While several different genera and species of P. aeruginosa prophages have been studied, there has not been a comprehensive study of the overall diversity of P. aeruginosa-infecting prophages. Here, we present the results of just such an analysis. A total of 6,852 high-confidence prophages were identified from 5,383 P. aeruginosa genomes from strains isolated from the human body and other environments. In total, 3,201 unique prophage sequences were identified. While 53.1% of these prophage sequences displayed sequence similarity to publicly available phage genomes, novel and highly mosaic prophages were discovered. Among these prophages, there is extensive diversity, including diversity within the functionally conserved integrase and C repressor coding regions, two genes responsible for prophage entering and persisting through the lysogenic life cycle. Analysis of integrase, C repressor, and terminase coding regions revealed extensive reassortment among P. aeruginosa prophages. This catalog of P. aeruginosa prophages provides a resource for future studies into the evolution of the species. IMPORTANCE Prophages play a critical role in the evolution of their host species and can also contribute to the virulence and fitness of pathogenic species. Here, we conducted a comprehensive investigation of prophage sequences from 5,383 publicly available Pseudomonas aeruginosa genomes from human as well as environmental isolates. We identified a diverse population of prophages, including tailed phages, inoviruses, and microviruses; 46.9% of the prophage sequences found share no significant sequence similarity with characterized phages, representing a vast array of novel P. aeruginosa-infecting phages. Our investigation into these prophages found substantial evidence of reassortment. In producing this, the first catalog of P. aeruginosa prophages, we uncovered both novel prophages as well as genetic content that have yet to be explored.
Keywords: Pseudomonas aeruginosa; prophages; temperate phages.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
Targeted deletion of Pf prophages from diverse Pseudomonas aeruginosa isolates has differential impacts on quorum sensing and virulence traits.J Bacteriol. 2024 May 23;206(5):e0040223. doi: 10.1128/jb.00402-23. Epub 2024 Apr 30. J Bacteriol. 2024. PMID: 38687034 Free PMC article.
-
Phage Morons Play an Important Role in Pseudomonas aeruginosa Phenotypes.J Bacteriol. 2018 Oct 23;200(22):e00189-18. doi: 10.1128/JB.00189-18. Print 2018 Nov 15. J Bacteriol. 2018. PMID: 30150232 Free PMC article.
-
A Temperate Sinorhizobium Phage, AP-16-3, Closely Related to Phage 16-3: Mosaic Genome and Prophage Analysis.Viruses. 2023 Aug 6;15(8):1701. doi: 10.3390/v15081701. Viruses. 2023. PMID: 37632043 Free PMC article.
-
Importance of prophages to evolution and virulence of bacterial pathogens.Virulence. 2013 Jul 1;4(5):354-65. doi: 10.4161/viru.24498. Epub 2013 Apr 23. Virulence. 2013. PMID: 23611873 Free PMC article. Review.
-
An overview on Vibrio temperate phages: Integration mechanisms, pathogenicity, and lysogeny regulation.Microb Pathog. 2022 Apr;165:105490. doi: 10.1016/j.micpath.2022.105490. Epub 2022 Mar 17. Microb Pathog. 2022. PMID: 35307601 Review.
Cited by
-
Hot Spots of Site-Specific Integration into the Sinorhizobium meliloti Chromosome.Int J Mol Sci. 2024 Sep 27;25(19):10421. doi: 10.3390/ijms251910421. Int J Mol Sci. 2024. PMID: 39408745 Free PMC article.
-
The role of filamentous matrix molecules in shaping the architecture and emergent properties of bacterial biofilms.Biochem J. 2024 Feb 21;481(4):245-263. doi: 10.1042/BCJ20210301. Biochem J. 2024. PMID: 38358118 Free PMC article. Review.
-
Gut phageome: challenges in research and impact on human microbiota.Front Microbiol. 2024 Mar 22;15:1379382. doi: 10.3389/fmicb.2024.1379382. eCollection 2024. Front Microbiol. 2024. PMID: 38585689 Free PMC article. Review.
-
Comprehensive deciphering prophages in genus Acetobacter on the ecology, genomic features, toxin-antitoxin system, and linkage with CRISPR-Cas system.Front Microbiol. 2022 Aug 2;13:951030. doi: 10.3389/fmicb.2022.951030. eCollection 2022. Front Microbiol. 2022. PMID: 35983328 Free PMC article.
-
Pseudomonas aeruginosa maintains an inducible array of novel and diverse prophages over lengthy persistence in cystic fibrosis lungs.FEMS Microbiol Lett. 2025 Jan 10;372:fnaf017. doi: 10.1093/femsle/fnaf017. FEMS Microbiol Lett. 2025. PMID: 39890605 Free PMC article.
References
-
- Casjens S, Hendrix RW. 2004. Bacteriophages and the bacterial genome, p 39–52. In The Bacterial Chromosome. John Wiley & Sons, Ltd, New York, NY.
-
- Little JW. 2005. Lysogeny, prophage induction, and lysogenic conversion, p 37–54. In Phages. John Wiley & Sons, Ltd, New York, NY.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
