

ATP-Sensitive Potassium Channels in Hyperinsulinism and Type 2 Diabetes: Inconvenient Paradox or New Paradigm?
- PMID: 35196393
- PMCID: PMC8893938
- DOI: 10.2337/db21-0755
ATP-Sensitive Potassium Channels in Hyperinsulinism and Type 2 Diabetes: Inconvenient Paradox or New Paradigm?
Abstract
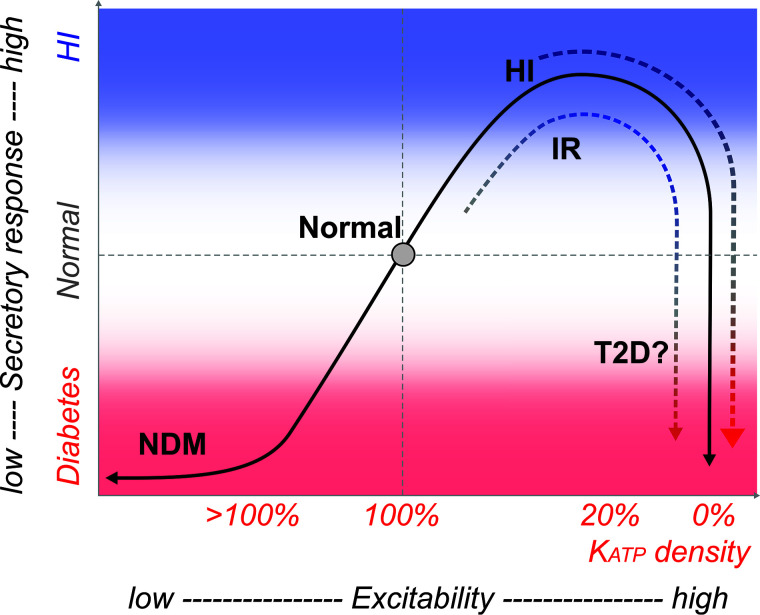
Secretion of insulin from pancreatic β-cells is complex, but physiological glucose-dependent secretion is dominated by electrical activity, in turn controlled by ATP-sensitive potassium (KATP) channel activity. Accordingly, loss-of-function mutations of the KATP channel Kir6.2 (KCNJ11) or SUR1 (ABCC8) subunit increase electrical excitability and secretion, resulting in congenital hyperinsulinism (CHI), whereas gain-of-function mutations cause underexcitability and undersecretion, resulting in neonatal diabetes mellitus (NDM). Thus, diazoxide, which activates KATP channels, and sulfonylureas, which inhibit KATP channels, have dramatically improved therapies for CHI and NDM, respectively. However, key findings do not fit within this simple paradigm: mice with complete absence of β-cell KATP activity are not hyperinsulinemic; instead, they are paradoxically glucose intolerant and prone to diabetes, as are older human CHI patients. Critically, despite these advances, there has been little insight into any role of KATP channel activity changes in the development of type 2 diabetes (T2D). Intriguingly, the CHI progression from hypersecretion to undersecretion actually mirrors the classical response to insulin resistance in the progression of T2D. In seeking to explain the progression of CHI, multiple lines of evidence lead us to propose that underlying mechanisms are also similar and that development of T2D may involve loss of KATP activity.
© 2022 by the American Diabetes Association.
Figures
References
-
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 1989;54:87–143 - PubMed
-
- Henwood MJ, Kelly A, Macmullen C, et al. . Genotype-phenotype correlations in children with congenital hyperinsulinism due to recessive mutations of the adenosine triphosphate-sensitive potassium channel genes. J Clin Endocrinol Metab 2005;90:789–794 - PubMed
-
- Remedi MS, Rocheleau JV, Tong A, et al. . Hyperinsulinism in mice with heterozygous loss of K(ATP) channels. Diabetologia 2006;49:2368–2378 - PubMed
-
- Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted overactivity of beta cell K(ATP) channels induces profound neonatal diabetes. Cell 2000;100:645–654 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
