

Low-dose metformin targets the lysosomal AMPK pathway through PEN2
- PMID: 35197629
- PMCID: PMC8891018
- DOI: 10.1038/s41586-022-04431-8
Low-dose metformin targets the lysosomal AMPK pathway through PEN2
Abstract
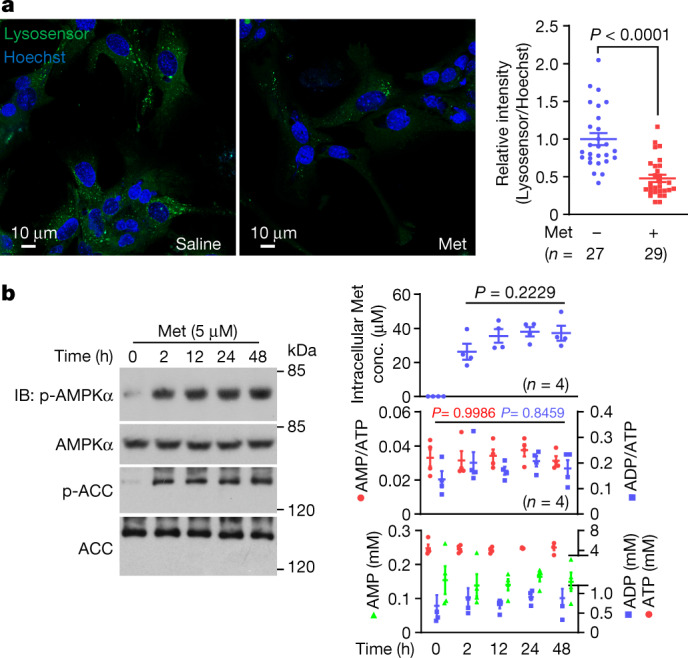
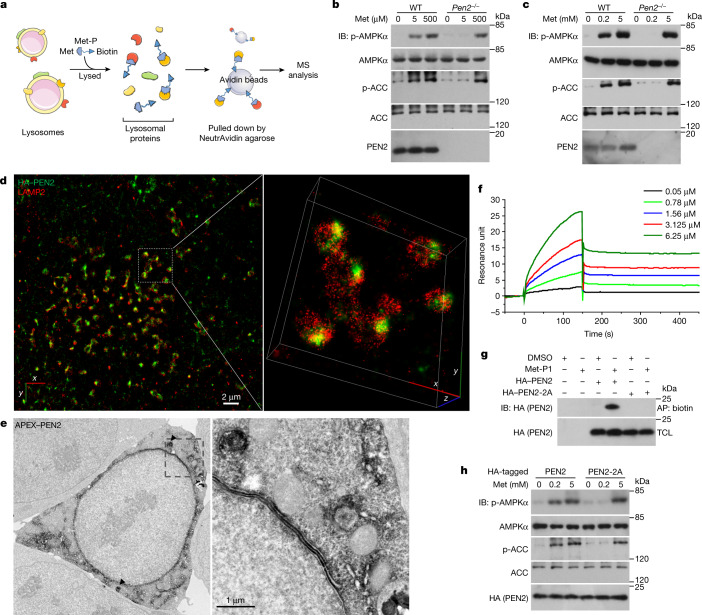
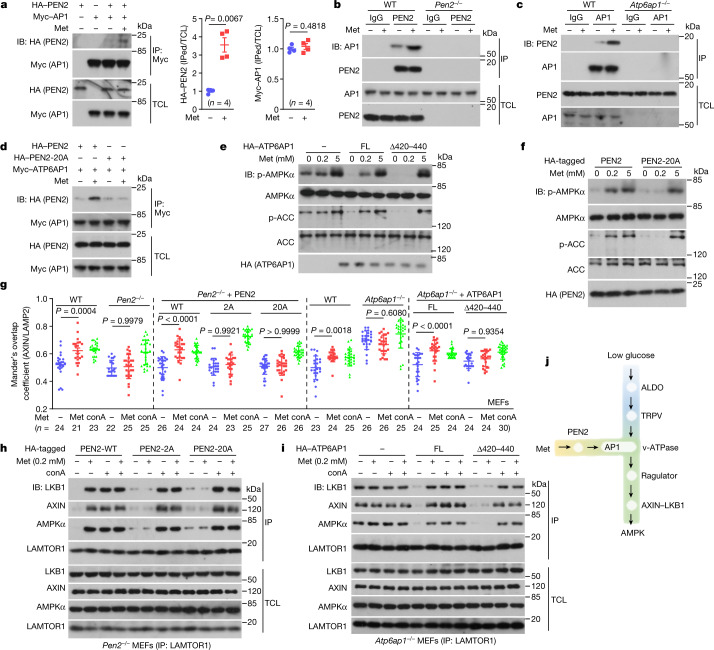
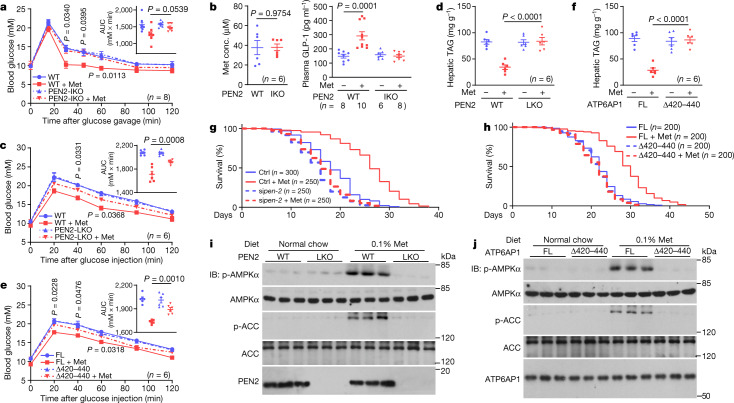
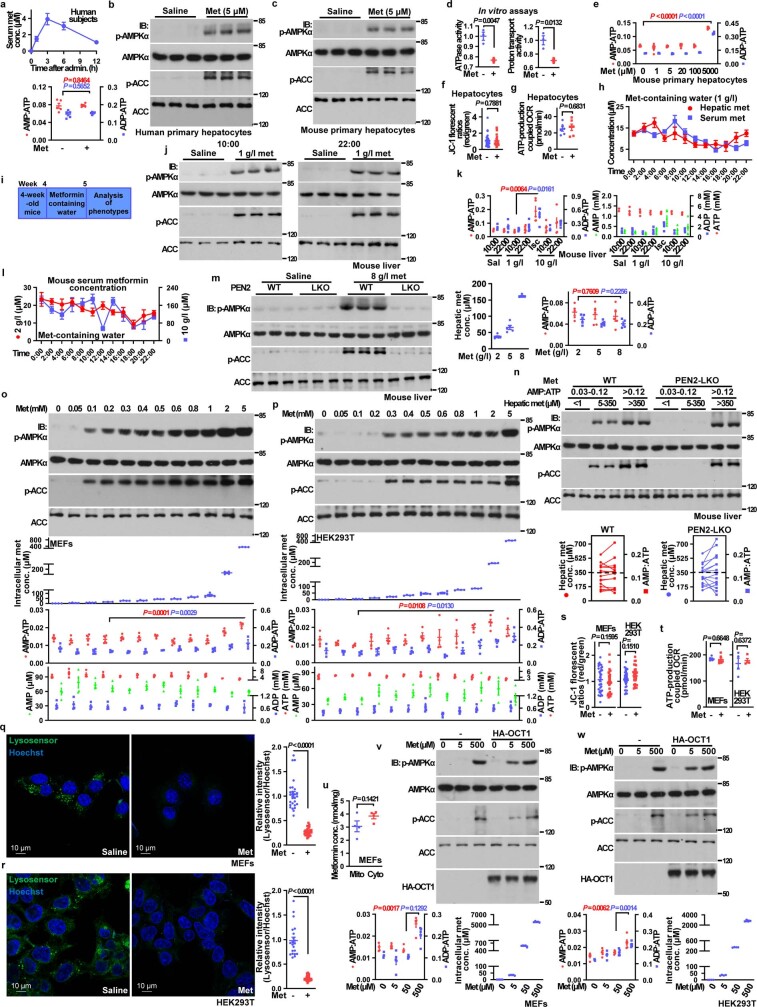
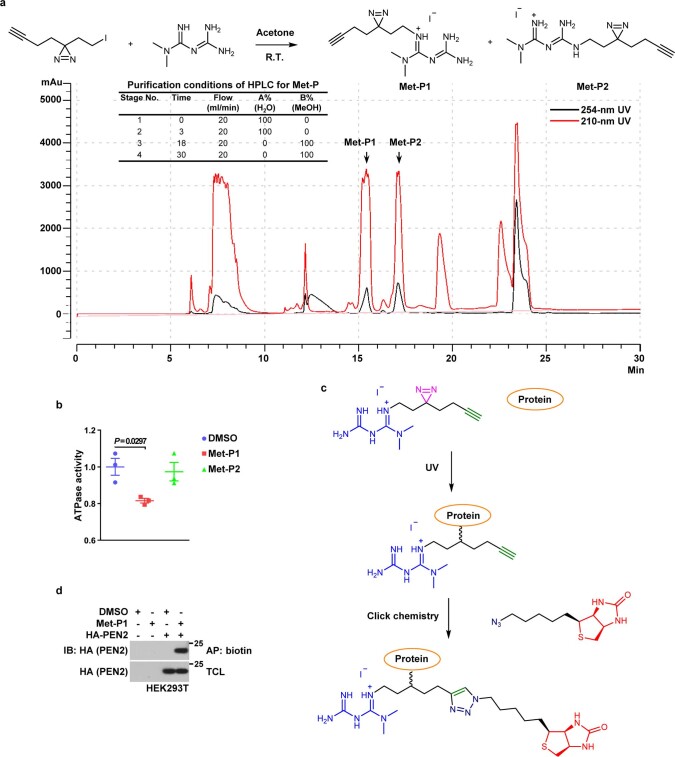
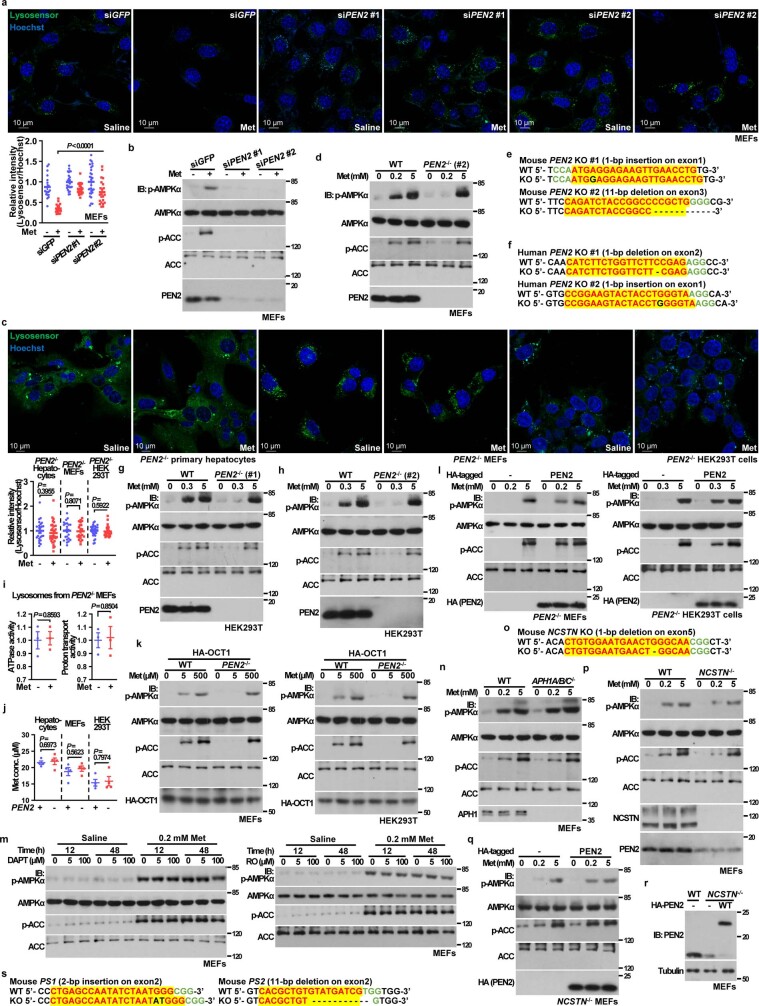
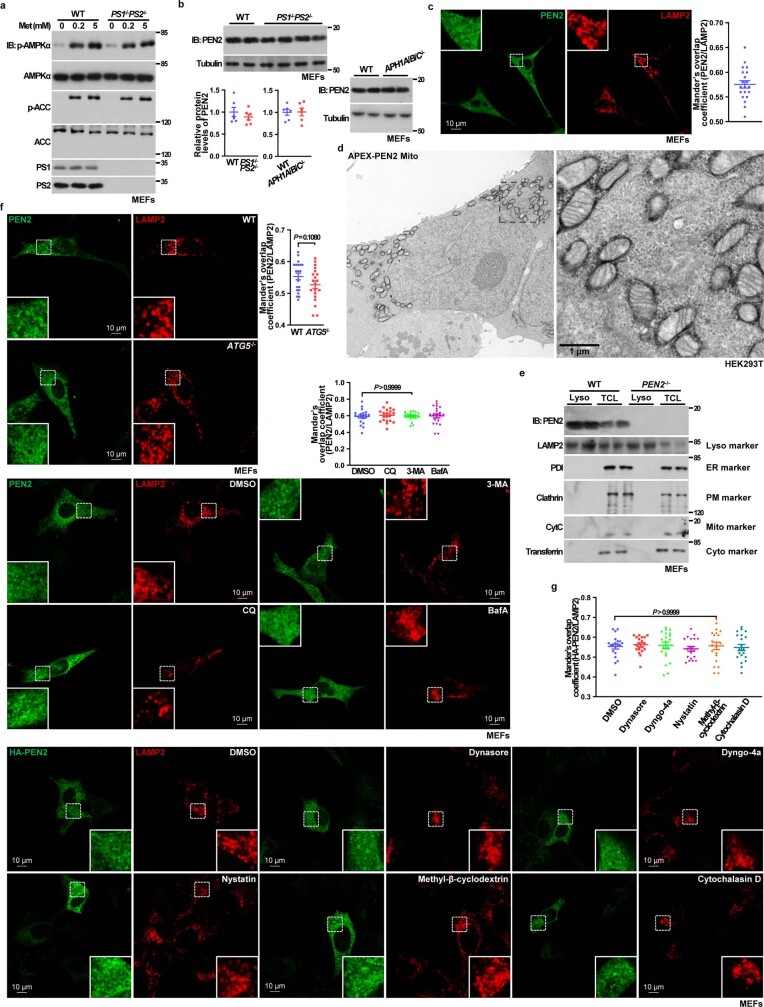
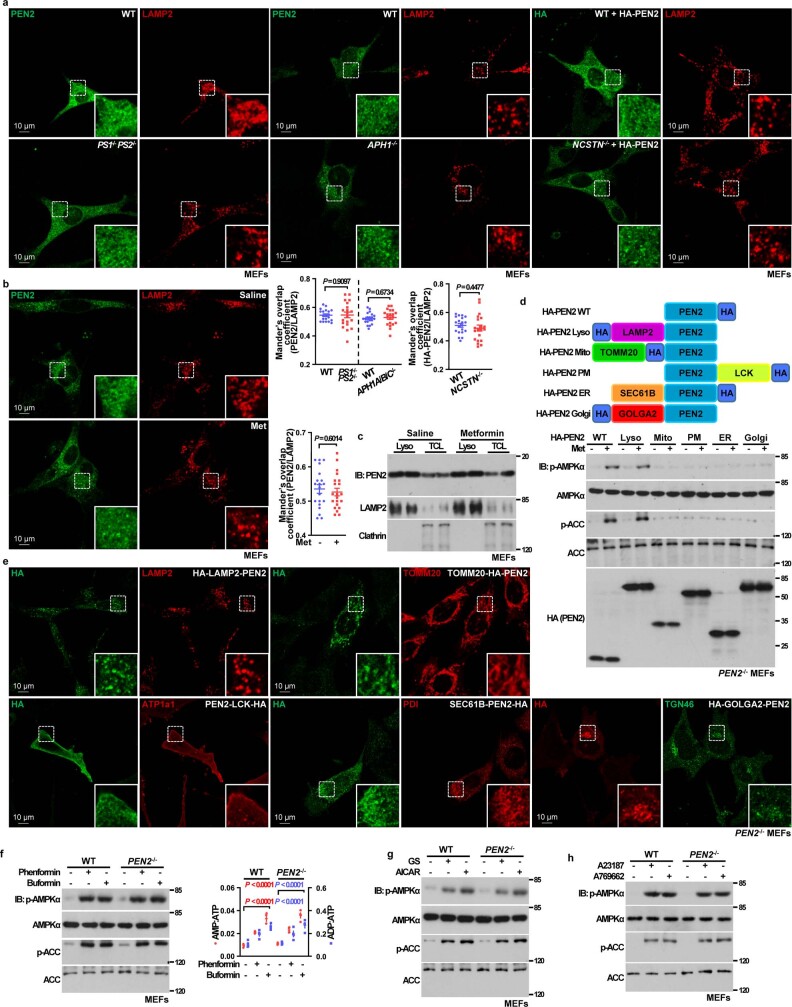
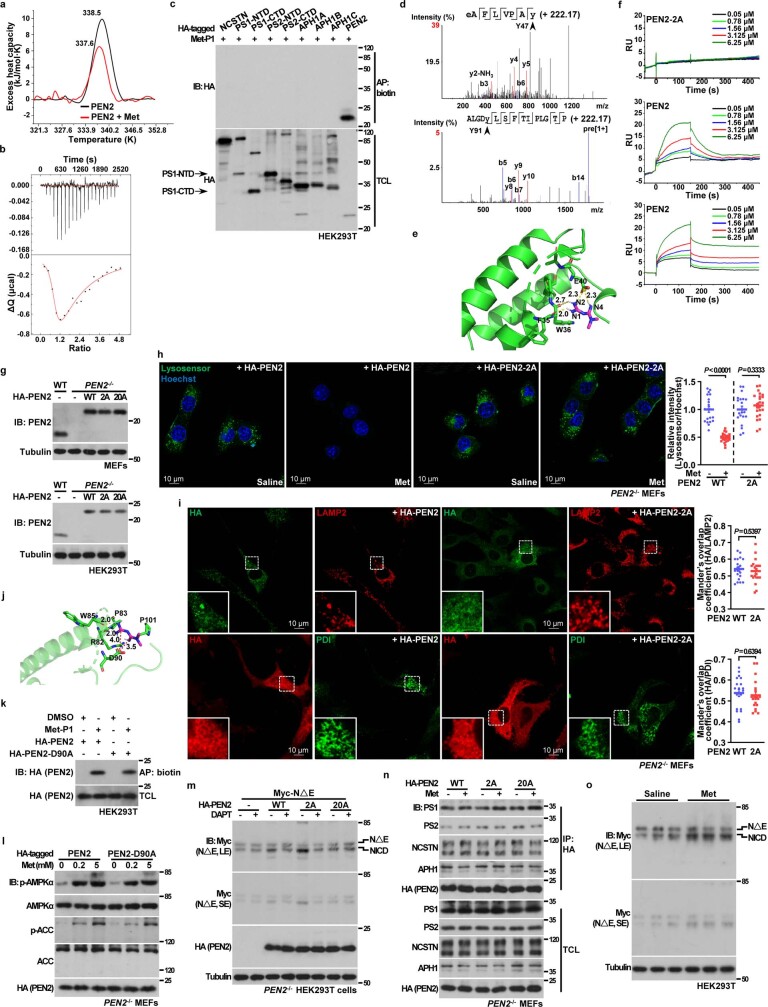
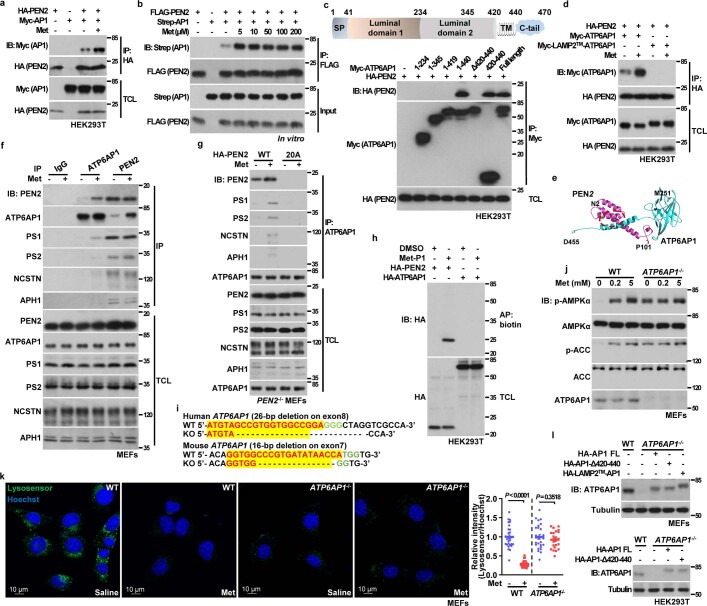
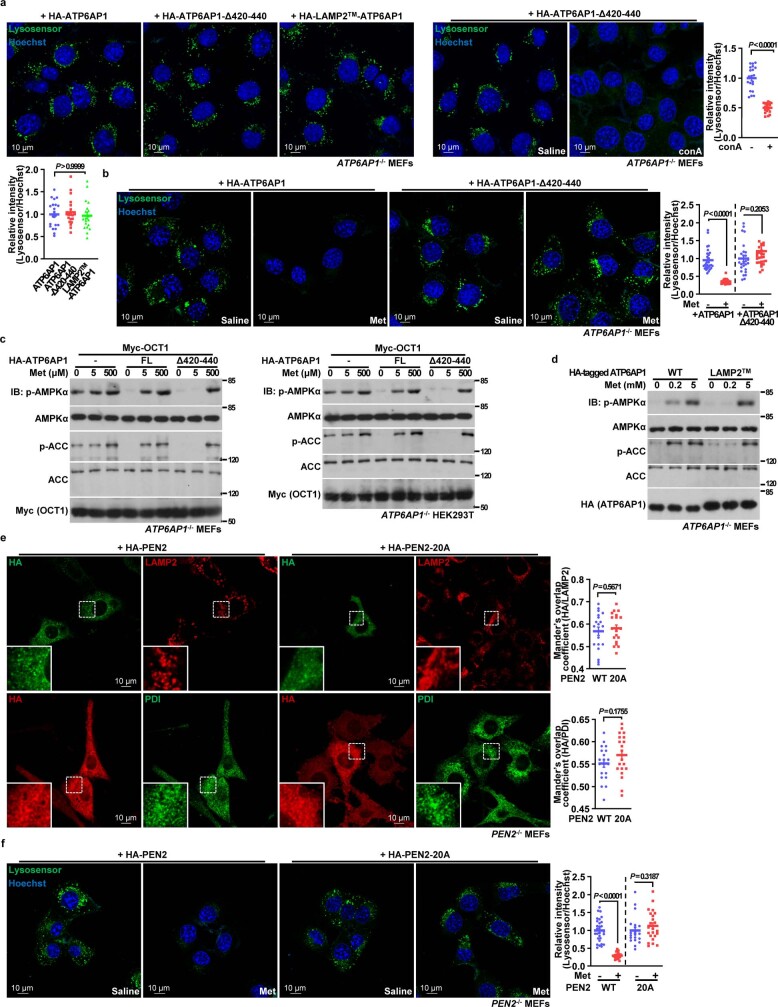
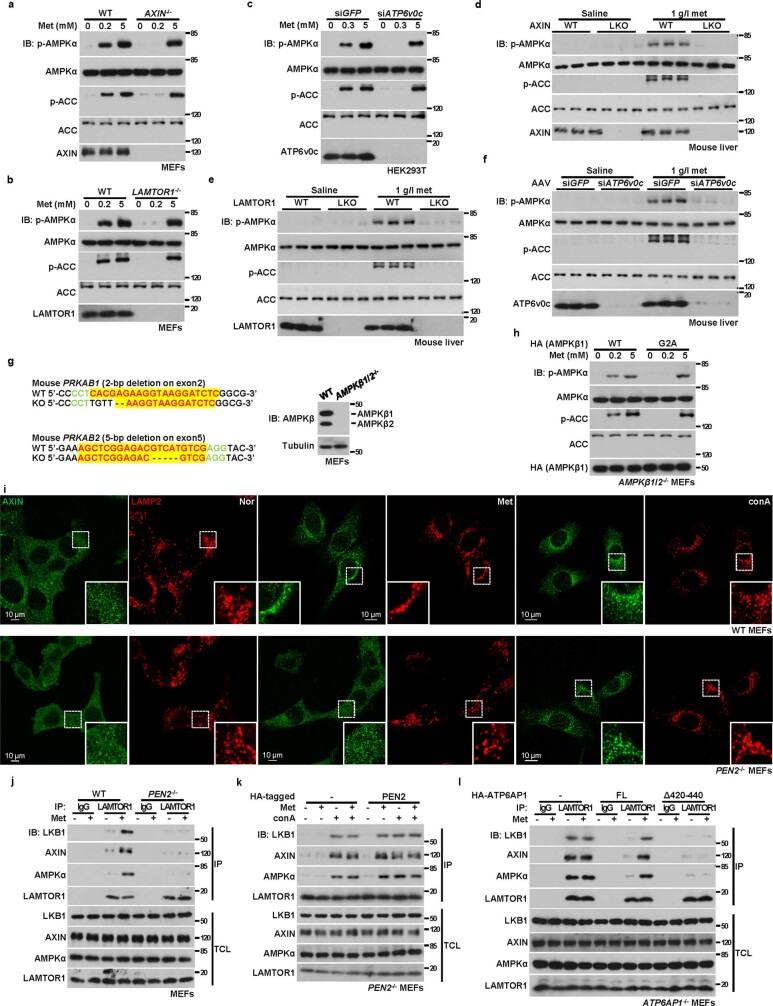
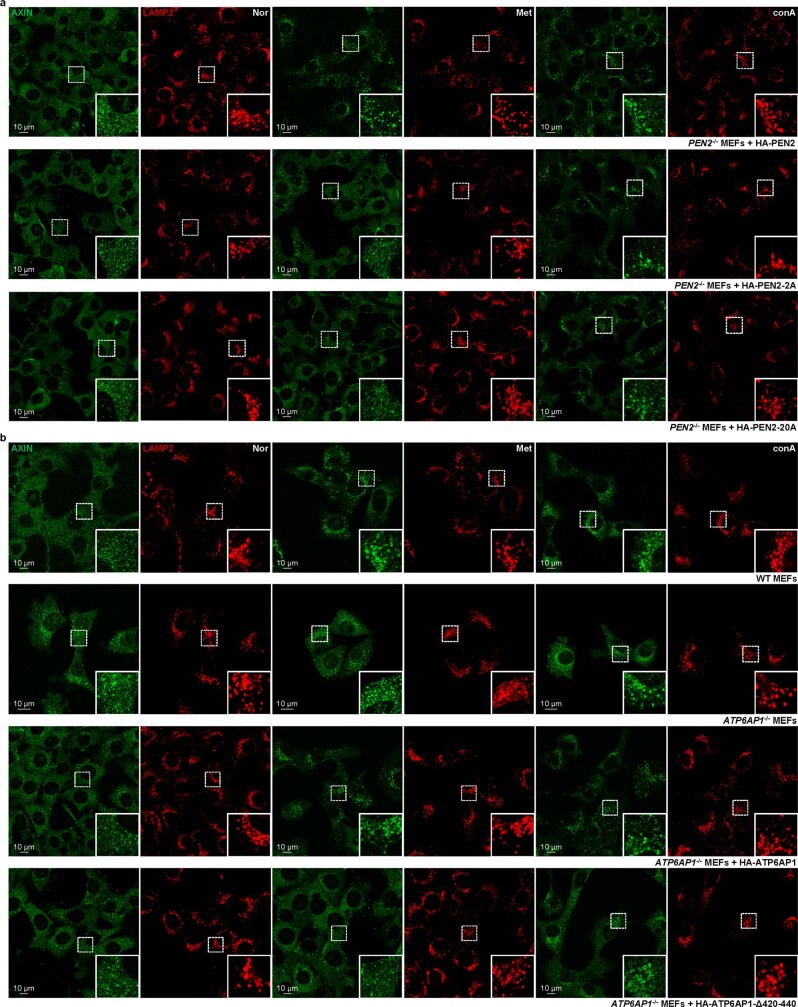
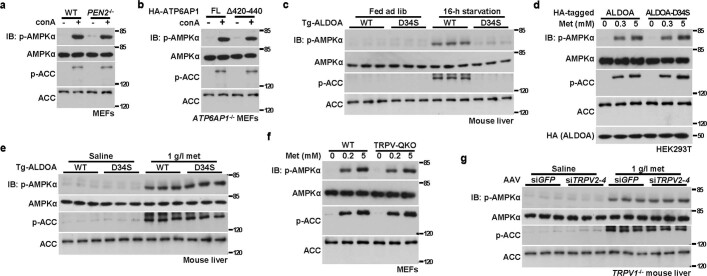
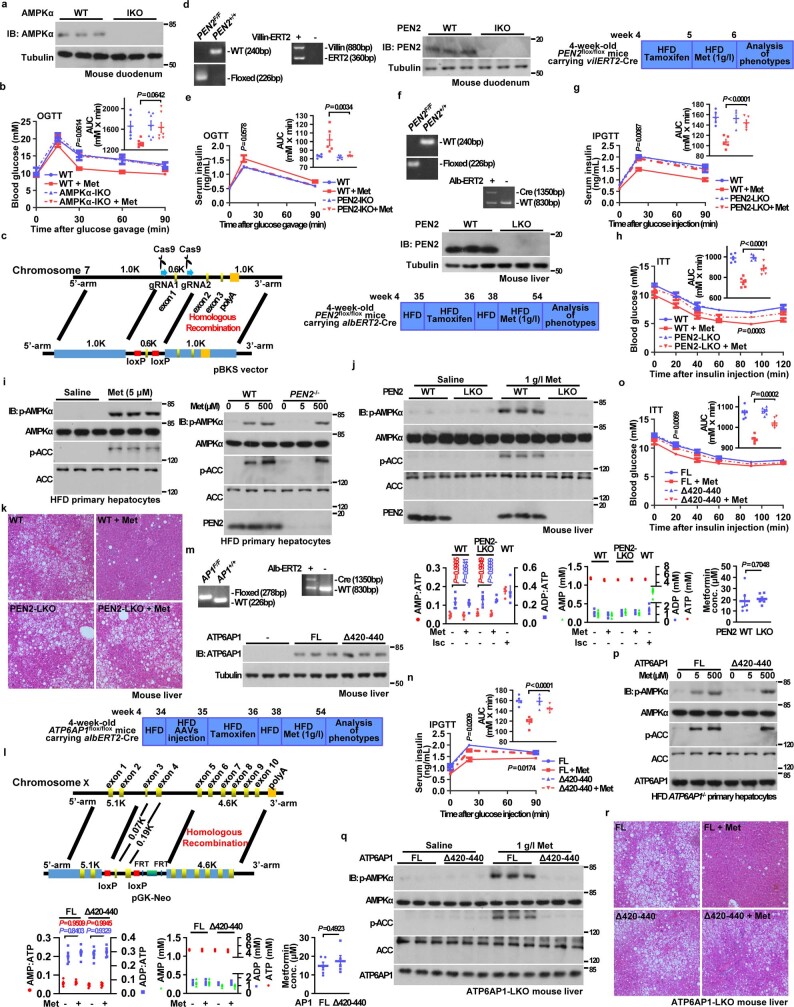
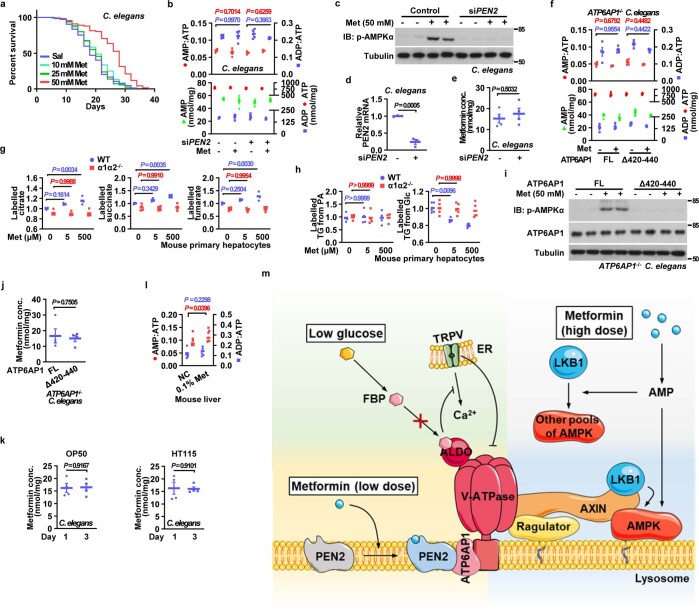
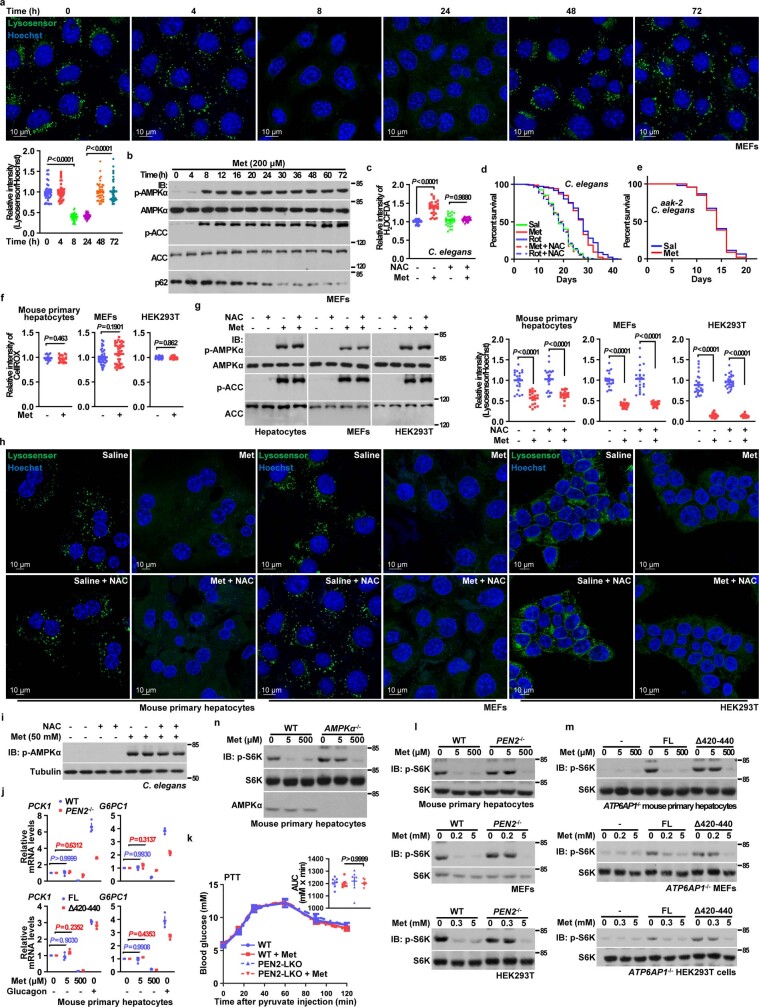
Metformin, the most prescribed antidiabetic medicine, has shown other benefits such as anti-ageing and anticancer effects1-4. For clinical doses of metformin, AMP-activated protein kinase (AMPK) has a major role in its mechanism of action4,5; however, the direct molecular target of metformin remains unknown. Here we show that clinically relevant concentrations of metformin inhibit the lysosomal proton pump v-ATPase, which is a central node for AMPK activation following glucose starvation6. We synthesize a photoactive metformin probe and identify PEN2, a subunit of γ-secretase7, as a binding partner of metformin with a dissociation constant at micromolar levels. Metformin-bound PEN2 forms a complex with ATP6AP1, a subunit of the v-ATPase8, which leads to the inhibition of v-ATPase and the activation of AMPK without effects on cellular AMP levels. Knockout of PEN2 or re-introduction of a PEN2 mutant that does not bind ATP6AP1 blunts AMPK activation. In vivo, liver-specific knockout of Pen2 abolishes metformin-mediated reduction of hepatic fat content, whereas intestine-specific knockout of Pen2 impairs its glucose-lowering effects. Furthermore, knockdown of pen-2 in Caenorhabditis elegans abrogates metformin-induced extension of lifespan. Together, these findings reveal that metformin binds PEN2 and initiates a signalling route that intersects, through ATP6AP1, the lysosomal glucose-sensing pathway for AMPK activation. This ensures that metformin exerts its therapeutic benefits in patients without substantial adverse effects.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
PEN2: Metformin's new partner at lysosome.Cell Res. 2022 Jun;32(6):507-508. doi: 10.1038/s41422-022-00661-7. Cell Res. 2022. PMID: 35418220 Free PMC article. No abstract available.
-
Low-dose metformin and PEN2-dependent lysosomal AMPK activation: benefits outnumber side effects.Signal Transduct Target Ther. 2022 Jun 4;7(1):178. doi: 10.1038/s41392-022-01040-9. Signal Transduct Target Ther. 2022. PMID: 35661690 Free PMC article. No abstract available.
References
-
- Foretz M, Guigas B, Viollet B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019;15:569–589. - PubMed
-
- Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015;66:17–29. - PubMed
-
- Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
