

Aldosterone-Regulated Sodium Transport and Blood Pressure
- PMID: 35197862
- PMCID: PMC8859437
- DOI: 10.3389/fphys.2022.770375
Aldosterone-Regulated Sodium Transport and Blood Pressure
Abstract
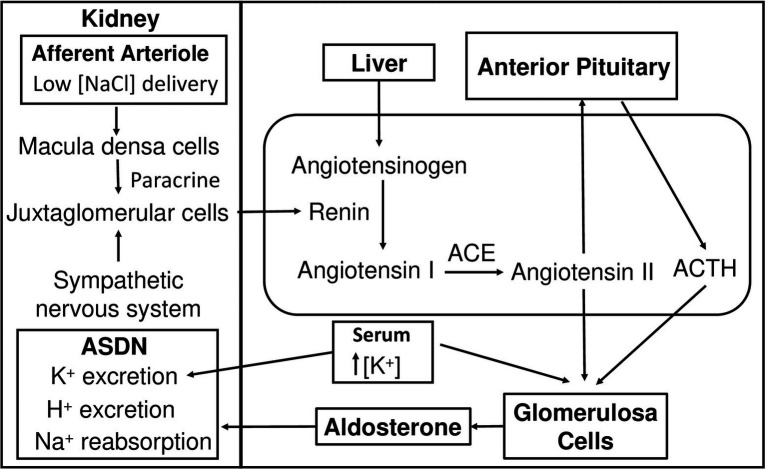
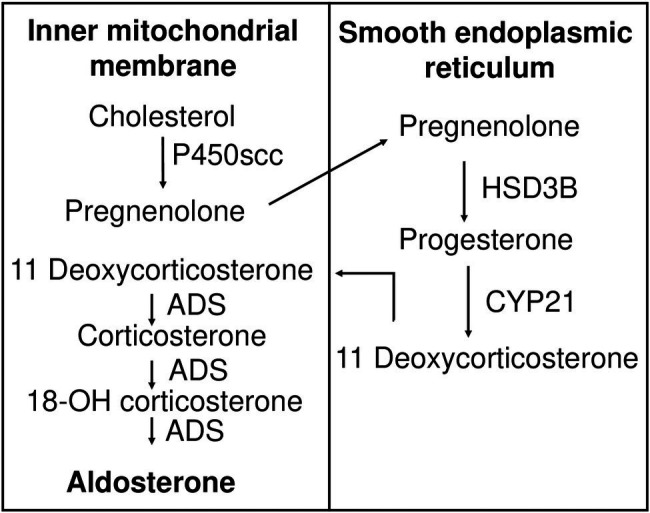
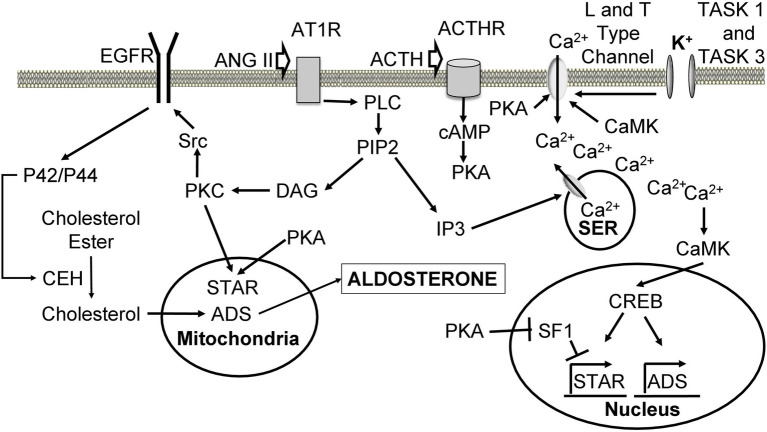
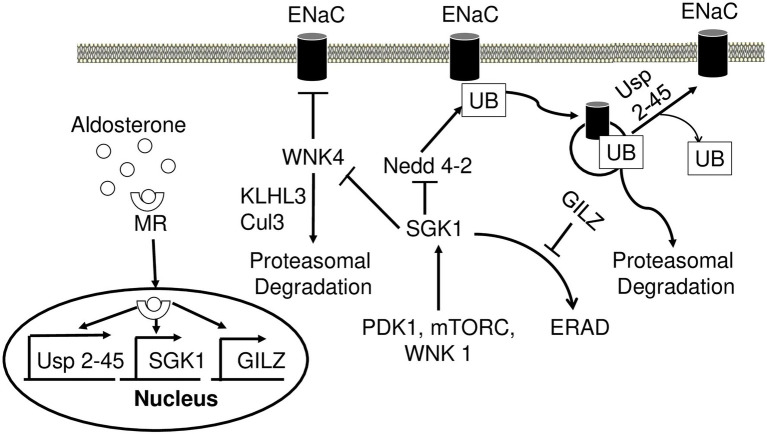
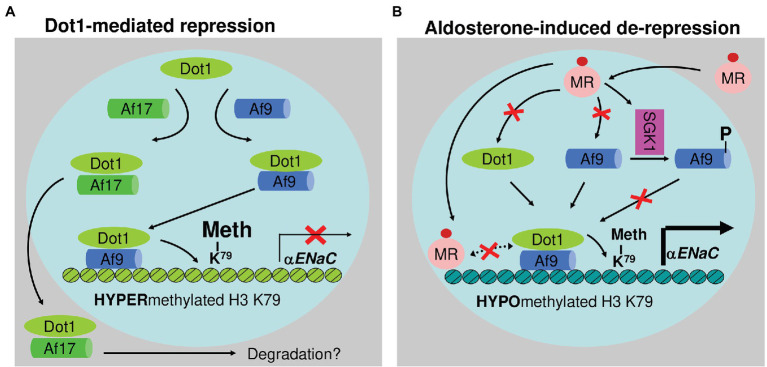
Aldosterone is a major mineralocorticoid steroid hormone secreted by glomerulosa cells in the adrenal cortex. It regulates a variety of physiological responses including those to oxidative stress, inflammation, fluid disruption, and abnormal blood pressure through its actions on various tissues including the kidney, heart, and the central nervous system. Aldosterone synthesis is primarily regulated by angiotensin II, K+ concentration, and adrenocorticotrophic hormone. Elevated serum aldosterone levels increase blood pressure largely by increasing Na+ re-absorption in the kidney through regulating transcription and activity of the epithelial sodium channel (ENaC). This review focuses on the signaling pathways involved in aldosterone synthesis and its effects on Na+ reabsorption through ENaC.
Keywords: ACTH; CYP11B2; Dot1; ENaC; SGK1; aldosterone; angiotensin II; potassium.
Copyright © 2022 Tsilosani, Gao and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Anderson J. V., Struthers A. D., Payne N. N., Slater J. D., Bloom S. R. (1986). Atrial natriuretic peptide inhibits the aldosterone response to angiotensin II in man. Clin. Sci. 70, 507–512. - PubMed
-
- Arriza J. L., Weinberger C., Cerelli G., Glaser T. M., Handelin B. L., Housman D. E., et al. (1987). Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237, 268–275. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources