

Type-I Interferon Signaling in Fanconi Anemia
- PMID: 35198459
- PMCID: PMC8859461
- DOI: 10.3389/fcimb.2022.820273
Type-I Interferon Signaling in Fanconi Anemia
Abstract
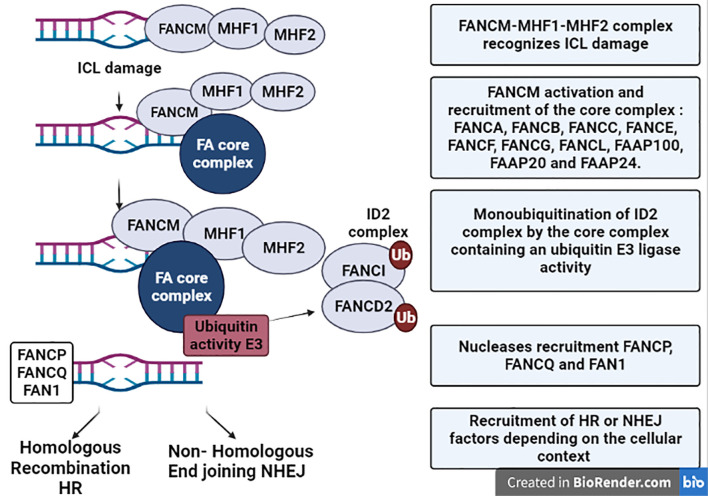
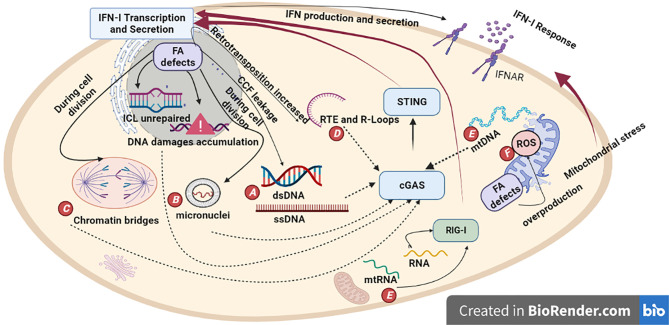
Fanconi Anemia (FA) is a genome instability syndrome caused by mutations in one of the 23 repair genes of the Fanconi pathway. This heterogenous disease is usually characterized by congenital abnormalities, premature ageing and bone marrow failure. FA patients also show a high predisposition to hematological and solid cancers. The Fanconi pathway ensures the repair of interstrand crosslinks (ICLs) DNA damage. Defect in one of its proteins prevents functional DNA repair, leading to the accumulation of DNA breaks and genome instability. Accumulating evidence has documented a close relationship between genome instability and inflammation, including the production of type-I Interferon. In this context, type-I Interferon is produced upon activation of pattern recognition receptors by nucleic acids including by the cyclic GMP-AMP synthase (cGAS) that detects DNA. In mouse models of diseases displaying genome instability, type-I Interferon response is responsible for an important part of the pathological symptoms, including premature aging, short stature, and neurodegeneration. This is illustrated in mouse models of Ataxia-telangiectasia and Aicardi-Goutières Syndrome in which genetic depletion of either Interferon Receptor IFNAR, cGAS or STING relieves pathological symptoms. FA is also a genetic instability syndrome with symptoms such as premature aging and predisposition to cancer. In this review we will focus on the different molecular mechanisms potentially leading to type-I Interferon activation. A better understanding of the molecular mechanisms engaging type-I Interferon signaling in FA may ultimately lead to the discovery of new therapeutic targets to rescue the pathological inflammation and premature aging associated with Fanconi Anemia.
Keywords: DNA damage; Fanconi anemia; RIG-I; cGAS/STING; cytosolic DNA; inflammation; interferon.
Copyright © 2022 Landelouci, Sinha and Pépin.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials